 ||]Clearly there is a need to be able to compare SMILES strings across different servers, canonical or not. Jmol accomplishes this using the find() function, which can compare structures with structures, structures with SMILES strings, and SMILES string with SMILES strings.||matching student-drawn structures|x = Jmol.evaluateVar(jmolApplet0,"\'" + stringAnswer + "\'.find("\'SMILES\',\'" + stringKey + "\') > 0")|JavaScript call to a Jmol applet to find out if a given SMILES string that a student has entered using a 2D drawing object matches the key. If required stereochemistry indicated in the key is missing in the answer, the result will be FALSE. If the student\'s answer has unnecessary stereochemistry indicated, the result will be TRUE.||]
||]Clearly there is a need to be able to compare SMILES strings across different servers, canonical or not. Jmol accomplishes this using the find() function, which can compare structures with structures, structures with SMILES strings, and SMILES string with SMILES strings.||matching student-drawn structures|x = Jmol.evaluateVar(jmolApplet0,"\'" + stringAnswer + "\'.find("\'SMILES\',\'" + stringKey + "\') > 0")|JavaScript call to a Jmol applet to find out if a given SMILES string that a student has entered using a 2D drawing object matches the key. If required stereochemistry indicated in the key is missing in the answer, the result will be FALSE. If the student\'s answer has unnecessary stereochemistry indicated, the result will be TRUE.||] @TILDEc@TILDE[MAN][MAN][MAN].@TILDEc@TILDE[MAN][MAN].@TILDEc@TILDE[MAN][MAN][MAN] ||generating a Jmol BioSMILES string with crosslinking|x = {{selected}}.find("SEQUENCE", true)|returns the Jmol bioSMILES sequence for the specified atoms (same as .find("SMILES",true), but without the header comment; "SEQ" here would give no comments at all.) Adding the optional second parameter TRUE adds crosslinking (for 1CRN):
//* chain A protein 1 *//@TILDEp@TILDETTC:1C:2PSIVARSNFNVC:3RLPGTPEAIC:3ATYTGC:2IIIPGA||generating a Jmol BioSMILES string with hydrogen bonding|x = {{selected}}.find("SEQUENCE", "H")|returns the Jmol bioSMILES sequence for the specified atoms with pre-calculated hydrogen bonds indicated as SMILES connection. After load =1crn;calculate HBONDS:
TC:1PGDYAN //* 46 *//
//* chain A protein 1 *// ˜p˜T:1TC:2:3:4C:5:6:7PS:8I:9V:%10A:%11R:8:%12S:9:%13
N:%10:%14F:%11:%15N:%12:%16V:%13:%17C:%14:%18R:%15:%19L:%17:%16PG:%19TP:%20:%21
E:%22A:%23I:%20:%24C:%21:%25:%18A:%22:%26T:%23Y:%24T:%25G:%26C:7I:3:2II:1
PGATC:4P:%27GDY:%27AN:6:5 //* 46 *// ||]
Jmol PolySMILES allows searching polyhedral faces of inorganic compounds or crystal structures rather than bond networks. The search is a local search, just of the atoms in the coordination sphere of a central atom. The SMILES string includes a "PH" chirality indicator, such as [Ru@PH7] for the central atom, specifing the number of attached atoms forming the corners of the polyhedron. the next n atoms (7 in this case) are appended using SMILES "." notation as though not connected to the central atom, instead showing connections to all associated edge atoms of that polyhedron. This listing is generated in a counterclockwise order looking toward the central atom from the specified corner. Jmol PolySMILES are generated by first creating polyhedra and then issuing calculate symmetry polyhedra. After this, a call to {{atomset}}.polyhedra.polySmiles gives the Jmol PolySMILES string.
load $P(Br)(Cl)(C)(O)(F);:>polyhedra 5;:>calculate symmetry polyhedra;:>print @1.polyhedra.polySmiles:>:>>[P@PH5].[F]1234.[O]5672.[C]8496.[Cl]185.[Br]379
Searching for Jmol PolySMILES involves {{*}}.polyhedra(@1.polyhedra.polySmiles), which in this case delivers ({{0}}), indicating that the first atom in the model (the phosphorus atom) matches this polyhedron.
This last example illustrates how we can invert the stereochemistry of the Jmol PolySMILES simply by changing "@PH5" to "@PH5@".
:>load $P(Br)(Cl)(C)(O)(F) :>polyhedra 5 :>calculate symmetry polyhedra :>x= @1.polyhedra.polySmiles :>print x :>:>>[P@PH5].[F]1234.[O]5672.[C]8496.[Cl]185.[Br]379:> :> // swap the axial ligands of the trigonal bipyramid (Br and Cl in this model) :>{{_Br}}.element = "B" :>{{_Cl}}.element = "Br" :>{{_B}}.element = "Cl" :>polyhedra delete :>polyhedra 5 :>calculate symmetry polyhedra :>print @1.polyhedra.polySmiles :>:>>[P@PH5].[F]1234.[O]5672.[C]8496.[Br]185.[Cl]379:> :>print {{*}}.polyhedra(x) :>:>>({{}}) // failed! :> :>print {{*}}.polyhedra(x.replace("@PH5","@PH5@")); // invert stereochemistry :>:>>({{0}}) // success! :> :>:> All the connections are the same, but all the winding is opposite -- the enantiomer. ','1|2','','') newCmd('.[JmolSQL]','','','*v+14.2;','Jmol maintains a large amount of information about atoms, bonds, shapes, files, and many other aspects of the models loaded into it. For example, getProperty("atomInfo") returns an array giving the information for each atom that is loaded:
:>load $caffeine :>x = getProperty("atomInfo") :>print x.count
:>>24
:>print x[1]
:>{{ :>> "_ipt" : 0 :>> "atomIndex" : 0 :>> "atomno" : 1 :>> "bondCount" : 3 :>> "clickabilityFlags" : 48 :>> "colix" : -32761 :>> "color" : "[x3050f8]" :>> "coord" : {{1.312 -1.0479 0.0025}} :>> "element" : "nitrogen" :>> "elemno" : 7 :>> "formalCharge" : 0 :>> "info" : "N1 #1" :>> "model" : "1" :>> "partialCharge" : 0.0 :>> "radius" : 0.7416667 :>> "shape" : "trigonal planar" :>> "spacefill" : 0.3565 :>> "sym" : "N" :>> "visibilityFlags" : 63 :>> "visible" : true :>> "x" : 1.312 :>> "y" : -1.0479 :>> "z" : 0.0025 :>}}
JmolSQL is a Jmol math syntax that is designed to query this sort of information. The idea is that maps, with key/value pairs, and especially arrays of maps, are data, and those arrays themselves can be thought of as a mini database. These sorts of data can be found in Jmol in the a model\'s auxiliary info (variable _M), including validation data returned from LOAD =xxxx/val (_M.validation), sequence domain data returned from LOAD =xxxx/dom (_M.domains), and secondary structure information returned from LOAD =xxxx/dssr (_M.dssr, see {misc/JmolSQLforDSSR.pdf~JmolSQLforDSSR.pdf}) or LOAD=xxxx/rna3d (_M.rna3d). In addition, the getProperty() function returns a wide variety of data relating to model attributes, including getProperty("atomInfo") and getProperty("bondInfo") among several others. (See {#.getProperty~}.) In addition, you can add your own database-like information to a model in the form of arrays and maps. The JmolSQL search query language syntax allows you to examine and analyze all of this information using a structured query language similar to MySQL. Any array or key/value paired "map" or "map" data can be interrogated.
The original conception of JmolSQL was an extension of the getProperty() function -- for example:
:>load $caffeine :>print getProperty("atomInfo[SELECT atomno,coord WHERE shape LIKE \'trigonal planar\']")
:> {{ :>> "atomno" : 1 :>> "coord" : {{1.312 -1.0479 0.0025}} :> }} :> {{ :>> "atomno" : 3 :>> "coord" : {{1.7906001 0.20809999 9.999999E-4}} :> }} :> ...24 of these...
The within(dssr,...) syntax also uses JmolSQL. See {misc/JmolSQLforDSSR.pdf~JmolSQLforDSSR.pdf} for details. The following selection finds and highlights RNA base pairs that are not standard Watson-Crick base pairs:
:>load =4fe5/dssr:>select on within(dssr, "pairs WHERE name!=\'WC\'");
More recent development widens this use to any array data, and use of the .select() function rather than getProperty() is recommended for general use. Thus, alternatively we can use:
:>print getProperty("atomInfo").select("atomno,coord WHERE shape LIKE \'trigonal planar\' ")
object.SELECT("keys WHERE/WHEREIN phrase")
There are three parts to JmolSQL: object, keys, and an optional WHERE or WHEREIN phrase. The object can be either a map [key1:value1, key2:value2, key3:value3] or, quite commonly, an array of maps, usually all having the same set of keys.
Maps
When the top-level object is a map, .select() can be used to select out subsets of that array, either as a single map or as an array of values.
map.select("...")
The simplest form of .select() returns a subset of map. Wild cards can be interspersed with additional keys, for example, "a*,b" or "*_id". In each such case, the case-sensitive LIKE operation is used to match keys.
:>map = [ A:[b:1],B:[b:2],AA:[b:3, d:50] ] :>print map.select("A").format("JSON")
:>>{{ "A": {{ "b": 1 }} }}
:>map = [ A:[b:1],B:[b:2],AA:[b:3, d:50] ] :>print map.select("A*").format("JSON")
:>>{{ "A": {{ "b": 1 }},"AA": {{ "b": 3,"d": 50 }} }}
map.select("(...)")
Using parentheses around the list of keys delivers an array of just the values rather than the full key/value pair:
:>map = [ A:[b:1],B:[b:2],AA:[b:3, d:50] ] :>print map.select("(A,B)").format("JSON")
:>>[ {{ "b": 1 }},{{ "b": 2 }} ]
Arrays of Maps
Properties such as AtomInfo, BondInfo, and ModelInfo are arrays of maps. A list of data for each atom, each bond, or each model. This is the essence of a database. JmolSQL can operate on these as well.
array.select("...")
JmolSQL will operate on an array of maps the same as on the map itself. But this time, the results will be in the form of an array of maps still. For example, we can create a sublist of maps having the selected subset of keys:
:>array = [ [a:1,b:11],[a:2,b:22],[a:3,b:33] ] :>print array.select("b").format("JSON")
:>>[ {{ "b": 11 }},{{ "b": 22 }},{{ "b": 33 }} ]
print x.select("atomno,element")
:>> {{ :>> "atomno" : 1 :>> "element" : "nitrogen" :>> }} :>> {{ :>> "atomno" : 2 :>> "element" : "carbon" :>> }} :>> {{ :>> "atomno" : 3 :>> "element" : "carbon" :>> }} :>> {{ :>> "atomno" : 4 :>> "element" : "oxygen" :>> }} :>> ...
Adding parentheses creates a list of only the values for the specified keys:
array = [ [a:1,b:11],[a:2,b:22],[a:3,b:33] ]
print array.select("(b)").format("JSON")
[ 11,22,33 ]
array = [ [a:1,b:11],[a:2,b:22],[a:3,b:33] ]
print array.select("(a,b)").format("JSON")
[ 11,1,22,2,33,3 ]
Let\'s catalog all the elements in a caffeine model:
load $caffeine
print getProperty("atomInfo").select("(element)").pivot
{{ :>> "carbon" : 8 :>> "hydrogen" : 10 :>> "nitrogen" : 4 :>> "oxygen" : 2 :>>}}
The assumption when using (keys) is that you want to know all these values, but you don\'t need the keys themselves, An example is a list of bonds for which we just want to know all the atoms involved, but the atoms are listed under "atom1" and "atom2" in each bond array.
load =1ehz/dssr
select on @{{_M.dssr.hbonds.select("(atom1_id,atom2_id)")}}
206 atoms selected
By the way, the reason we can go so directly from {http://dssr-jmol.x3dna.org/report.php?id=1ehz&opts=--json=ebi~DSSR data structures} to atom selections is that DSSR atom and residue descriptions use Unit IDs. The _M.dssr.hbonds structure looks like this:
:>{{:>> "atom1_id" : "|1|A|G|1|OP2|||":>> "atom1_serNum" : 4:>> "atom2_id" : "|1|A|C|2|OP2|||":>> "atom2_serNum" : 27:>> "atom_pair" : "O:O":>> "distance" : 2.991:>> "donAcc_type" : "questionable":>> "index" : 1:>> "res_long" : "|1|A|G|1||||,|1|A|C|2||||":>> "res_short" : "GC":>> "residue_pair" : "nt:nt" :> }}
Jmol can interpret Unit IDs natively, and, because Unit IDs have such a unique signature, Jmol can even select atoms based on any text that contains them. They do not have to be given individually.
Using WHERE
WHERE is used to select a subset of the elements of an array based on specific key-value relationships.
array.select("... WHERE ...")
Delivers all key/value pairs in the subset of array element maps for which the WHERE clause is true for that element.
:>array = [ [a:1,b:11],[a:2,b:22],[a:3,b:33] ] :>print array.select("* WHERE a<3 and b<20").format("JSON")
:>>[ {{ "b": 11,"a": 1 }} ]
:>load $caffeine :>print getProperty("atomInfo").select("atomno,element WHERE shape LIKE \'trigonal planar\' ").format("JSON")
:>>[ {{ "element": "nitrogen","atomno": 1 }},{{ "element": "carbon","atomno": 3 }},{{ "element": "nitrogen","atomno": 5 }},{{ "element": "carbon","atomno": 7 }},{{ "element": "carbon","atomno": 9 }},{{ "element": "nitrogen","atomno": 10 }},{{ "element": "carbon","atomno": 12 }},{{ "element": "carbon","atomno": 13 }} ]
array.select("(...) WHERE ...")
Using parentheses around the list of keys delivers a list of values for only the subset of array for which the WHERE clause is true:
:>array = [ [a:1,b:11],[a:2,b:22],[a:3,b:33] ] :>print array.select("(b) WHERE a>1").format("JSON")
:>>[ 22,33 ]
This next example quantifies the various atomic orbital hybridizations that in the caffeine molecule:
:>load $caffeine :>print getProperty("atomInfo").select("(shape) WHERE shape").pivot
:>{{ :>> "bent" : 1 :>> "tetrahedral" : 3 :>> "trigonal planar" : 8 :>}}
Note that "WHERE shape" here just excludes all cases where shape is the empty string, since empty strings in Jmol evaluate as FALSE. (In this case that involves hydrogen atoms.)
This next example finds all the hydrogen bonds created by DSSR for residue 72, listing and averaging them. Note that the DSSR report uses {https://www.bgsu.edu/research/rna/help/rna-3d-hub-help/unit-ids.html~Unit IDs} to represent residues:
:>load =1ehz/dssr :>x = _M.dssr.hbonds.select("(distance) WHERE res_long LIKE \'*|A|C|72|*\'"); :>print x.format("JSON") :>print format("%5.3f",x.average)
:>>[ 2.832,2.879,2.838 ] :>>2.850
Array Drilling
WHERE will "drill down" through arrays of arrays to find elements that are maps.
:>array = [ [[a:1,b:11], [a:0,b:0]],[[[a:2,b:22]]],[[a:3,b:33,aa:44]] ] :> print array.select("a* WHERE a>0").format("JSON")
:>>[ [ {{ "a": 1 }} ],[ [ {{ "a": 2 }} ] ],[ {{ "a": 3,"aa": 44 }} ] ]
We use array.join() here to produce a flat one-dimensional array of those objects:
:>array = [ [[a:1,b:11], [a:0,b:0]],[[[a:2,b:22]]],[[a:3,b:33,aa:44]] ] :>print array.select("a* WHERE a>0").join().format("JSON")
:>>[ {{ "a": 1 }},{{ "a": 2 }},{{ "a": 3,"aa": 44 }} ]
Note how effective array.join() is in this next case:
:>array = [ [[a:1,b:11], [a:0,b:0]],[[[a:2,b:22]]],[[a:3,b:33,aa:44]] ] :>print array.select("(b) WHERE a>0").join().format("JSON")
:>>[ 11,22,33 ]
Using WHEREIN
Using select(... WHEREIN...), you can select key value pairs where the values of the selected keys themselves are tested. For example:
:>x = [key_1:[type:"a"],key_2:[type:"b"],key_3:[type:"a"]]:>print x.select("* WHEREIN type=\'a\'").format("JSON")
:>{{ "key_1":{{ "type":"a" }},"key_3":{{ "type":"a" }} }}
Note that the resultant array now only has entries for key_1 and key_3, because only they have values for which type == "a". (Had we used WHERE here, nothing would be returned, because "type" is not a key in x itself.)
This next example checks the symmetry of occupied orbitals in Gausian output using WHEREIN:
:>load https://chemapps.stolaf.edu/jmol/jsmol/data/no2_nbo.log.gz 2 filter "alpha" :>print _M.moData.select("mos WHEREIN occupancy>0").select("(symmetry)").pivot
:>{{ :>> "(A1)--O" : 6 :>> "(A2)--O" : 1 :>> "(B1)--O" : 1 :>> "(B2)--O" : 4 :>}}
We needed to use WHEREIN here because the symmetry information is not in _M.moData itself; it is in _M.moData.mos. Admittedly, in this case, we could have accomplished this more directly with WHERE, using _M.moData.mos rather than _M.moData itself:
:>load https://chemapps.stolaf.edu/jmol/jsmol/data/no2_nbo.log.gz 2 filter "alpha" :>print _M.moData.mos.select("(symmetry) WHERE occupancy>0").pivot
:>{{ :>> "(A1)--O" : 6 :>> "(A2)--O" : 1 :>> "(B1)--O" : 1 :>> "(B2)--O" : 4 :>}}
This next example identifies all orbitals in a Gaussian output with B2 symmetry using WHEREIN:
:>load https://chemapps.stolaf.edu/jmol/jsmol/data/no2_nbo.log.gz 2 filter "alpha" :>x= _M.moData.select("mos WHEREIN occupancy>0 and symmetry LIKE \'(B2)*\' ") :>print x.select("(index)").format("JSON")
:>>[ 1,5,8,10 ]
chaining .select()
Note that JmolSQL can be chained:
:>x = getProperty("measurementInfo").select("(atoms) WHERE value > 1.4").select("(atomno)");
In this case, the first selection returns an array of atom records, and the second selection pulls out only the values for atomno, producing something such as [ [ 12,13 ],[ 2,1 ],[ 6,5 ],[ 11,10 ] ].
using select([keys])
Here is another example. It produces a listing of all atoms involving double bonds in the NCI/CADD caffeine model:
:>load $caffeine :>print getProperty("bondInfo").select("(atom1,atom2) WHERE type=\'double\'").select("(info)").format("JSON")
:>>[ "C3 #3","O4 #4","N8 #8","C9 #9","C7 #7","C12 #12","C13 #13","O14 #14" ]
This is OK, but wouldn\'t we want to have those paired by bonds? Changing (atom1,atom2) to [atom1,atom2] solves this issue:
:>load $caffeine :>print getProperty("bondInfo").select("[atom1,atom2] WHERE type=\'double\'").select("(info)").format("JSON")
:>>[ [ "C3 #3","O4 #4" ],[ "N8 #8","C9 #9" ],[ "C7 #7","C12 #12" ],[ "C13 #13","O14 #14" ] ]
The first SELECT finds all the double bonds, creating an array of [atom1,atom2] data. The second SELECT then replaces the full atom record with just the info value for each atom.
Thus, when working with arrays, whether SELECT returns just values or a full set of matching key:value pairs (as in standard SQL) is determined by how the keys are listed. If keys are simply listed, without [...] or (...), then what is returned is a flat array containing the specified keys with their values. Thus, array.select("nt1,nt2") creates an array containing maps that are a subset of array that contain keys nt1 and nt2 and their associated values. array.select("(nt1,nt2)") creates a single array that holds all matching values, and array.select("[nt2,nt1]") creates an array of arrays of [nt2,nt1] values. (By the way, we could use xxx.format(["nt2","nt1"]) to give the same result.) ','1|2','','') newCmd('.[Jmol/JSpecView/MagresView]','12.3.16','','*v+13.0;load;sync','Jmol 13.0
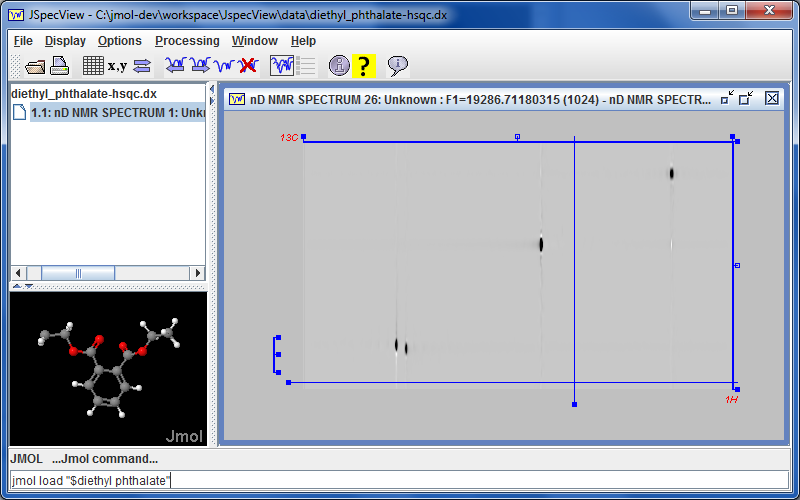 marks a new collaboration with the {jspecview.sourceforge.net~JSpecView project} with the incorporation of JSpecView into the Jmol application and synchronized communication between the Jmol applet and the JSpecView applet. This is most evident in that there is a new menu item Tools...JSpecView, which opens a new JSpecView frame that includes the current model. Work is in progress in the design and implementation of a new JCAMP-DX format that incorporates models and assignments into JDX files using custom ##$MODELS and ##$PEAKS records. See {misc/Jmol-JSpecView-specs.pdf~Jmol-JSpecView-specs.pdf} for proposed specification. Progress can be monitored at {http://chemapps.stolaf.edu/jmol/jspecview~}. The Jmol application command SYNC ON; sync * "JSpecView:..." sends commands to JSpecView.
marks a new collaboration with the {jspecview.sourceforge.net~JSpecView project} with the incorporation of JSpecView into the Jmol application and synchronized communication between the Jmol applet and the JSpecView applet. This is most evident in that there is a new menu item Tools...JSpecView, which opens a new JSpecView frame that includes the current model. Work is in progress in the design and implementation of a new JCAMP-DX format that incorporates models and assignments into JDX files using custom ##$MODELS and ##$PEAKS records. See {misc/Jmol-JSpecView-specs.pdf~Jmol-JSpecView-specs.pdf} for proposed specification. Progress can be monitored at {http://chemapps.stolaf.edu/jmol/jspecview~}. The Jmol application command SYNC ON; sync * "JSpecView:..." sends commands to JSpecView. Starting with Jmol 14.0, the Jmol application can access simulated NMR specta using the {#.show_NMR~show NMR} command and display them along with their correlations.
Also starting with Jmol 14.0, functionality is added for interacting with {http://www.ccpnc.ac.uk/pmwiki.php/CCPNC/MagresView~MagresView} in relation to calculation and visualization of solid-state NMR constants: [||{{*}}.ms and %[ms] (for labels) | magnetic shielding tensor isotropic values||{{*}}.cs and %[cs] (for labels) | chemical shift tensor isotropic values||set shift_XX xxx | sets chemical shift offset, in ppm||getProperty nmrInfo | delivers the table of isotopes|| print getproperty("nmrinfo.isotopes")["2H"] | an array for deuterium including [n,dc,qc] where n is the isotope number, (negative if the most abundant isotope), dc is the dipolar coupling constant, and qc is the quadrupolar coupling constant.||measure({{a}} {{b}} "isc_hz") | function returning Magres J-coupling value||measure({{a}} {{b}} "dc_khz") | function returning Magres dipolar constant|| MEASURE {{a}} {{b}} "2://dc_hz" | measurement line showing dipolar constant, Hz|| MEASURE {{a}} {{b}} "2:%3.2VALUE//dc_khz" | measurement line showing dipolar constant, kHz|| MEASURE {{a}} {{b}} "2://khz" | measurement defaults to dc_khz|| MEASURE {{a}} {{b}} "2://hz" | measurement defaults to isc_hz|| MEASURE {{a}} {{b}} "2://isc_1hz" | measurement line showing specifically "isc_1" values in Magres file|| {{xxx}}.tensor(type,what) | full access to tensor quantities, including j, chi, dc, eigenvalues, eigenvectors, value, asymMatrix, symMatrix, isotropy, anisotropy, asymmetry, eulerzyz, euelerzxz, quaternion, indices, type id, span, and skew.||]','1|2','','') newCmd('.[Jmol and Symmetry]','','','*v+13.0;load;unitcell;show','Jmol has a full set of analysis and visualization tools for space group and point group symmetry. All standard and magnetic space group operations are covered, including "3+N" higher-dimensional symmetry for incommensurately modulated crystal structures. Features are concentrated in four command options and one function:
:> LOAD .... SPACEGROUP .... UNITCELL ....:> SHOW SYMOP ....:> DRAW SYMOP .... :> DRAW POINTGROUP or SPACEGROUP:> x = symop(....)
With these features, you can:
-- load any model while applying any actual or conceivable space group or subset of a space group
-- tabulate and visualize all space group operations associated with that model
-- retrieve an associative array indicating all aspects of the operation, including point, rotation axis, plane, glide vector, lattice offset vector, and 4x4 matrix
-- visualize all aspects of an operation, including proper and improper rotations inversion centers, mirror lanes, glideplanes, translations, and magnetic spin time reversal
-- apply any operation to any atom or any point in space
-- given any combination of two atoms or two points in space, determine and/or depict the operation(s) that relate those to atoms or positions.','1|2','','') newCmd('.[Jmol and Chirality]','','','','Jmol has a relatively complete set of algorithms to assign {https://pubs.acs.org/doi/10.1021/acs.jcim.8b00324~Cahn-Ingold-Prelog} chirality descriptors, including R/S/r/s, E/Z, and M/P/m/p (axial chirality, such as for cumulenes and the single bonds between aromatic compounds with restricted rotation -- that is, {https://en.wikipedia.org/wiki/Atropisomer~atropisomerism}). There are several ways to access this information:[|| labeling| The LABEL and SET LABELFOR commands with format %[chirality] display the chirality of all selected atoms, if it exists. Bond chirality is shown at both ends. ||
The designations n and m are one of 1, 2, or 3, with n indicating the index of the bond for the atom on the left, and m the same for the atom on the right. In this case, the first "2" points to ring connection 1, connecting to nitrogen, and the second "2" points to the second connection to the carbon on the right, that is to ring connction 2, again a nitrogen. The defaults "^22-" and "^^22-" can be left simply as "^-" and "^^-".|| Jmol SMILES and SMARTS| You can use Jmol SMILES and SMARTS to query a structure and check its chirality. Several methods, particularly FIND and COMPARE and especially powerful in this regard, allowing all sorts of highly sophisticated analysis. ||]','1|2','','') newCmd('.[functions]','','','*v+14.2 -- adds support for private functions;math','Jmol math allows for two general syntaxes for functions, x = f(y) and x.f(y). The first simply operate on their given parameters. For example, x = load("myfile.dat") loads the variable x with the contents of the file "myfile.dat". The second operate on the elements of a variable individually in some way and are referred to here as item selector functions. In the listings below, these functions all begin with a period character. For example, x = {{carbon}}.bonds operates so as to deliver the bond set associated with the carbon atoms; x = "this is a test".replace("s","S") operates on the individual characters of the string "this is a test"; x = {{oxygen}}.label("%U") assigns x the list of labels for the oxygen atoms in the model. Some functions can be used in both contexts. For example, x = distance({{oxygen}}, {{carbon}}) delivers the distance from the CENTER of the oxygens to the CENTER of the carbons. x = {{oxygen}}.distance{{carbon}} delivers the AVERAGE distance of an oxygen atom to the CENTER of the carbons. These are subtly different. Some functions require parentheses; some do not. Basically, if a function CAN have parameters, for example, .join(), .split(), or .label(), then it MUST have at least empty (); if a function CANNOT have parameters, for example, .atoms, .bonds, or .ident, then it NEVER uses parentheses.','0|1','','') newCmd('','14.1.12','','*v+11.2;','Several of these functions are described more fully under the heading {#.atomexpressions~atom expressions}, as they can also be used in commands such as {#.select~} and {#.display~}.[|| x = abs(y)|the absolute value of y|| x = acos(y)|the arccosine of y, in degrees in the range 0 to 180 .||x = angle(a,b,c)|the a-b-c angle, where a, b, and c can be points or atom sets.||x = angle(a,b,c,d)| the dihedral angle a-b-c-d is measured.||x = cache()|returns an array describing the contents of the file {#.cache~} in the format {{filename: nBytes, filename: nbytes,...}} ||x = color(color1,color2,nColors,asHSL)| Returns a color scheme string spanning the distance between color1 and color2 with nColors colors. The last parameter, asHSL, if TRUE does the gradient in terms of hue, saturation, and lightness (HSL), which is better for cross-color gradients such as red-to-blue; if FALSE, uses RGB, which is better for single-color gradients such as white to red. The standard Jmol "rgb" color scheme is very close to color("red","blue",33,true). Once a variable is defined as a color scheme string, it can be used in commands such as {#.color~} property temperature @x and {#.isosurface~} vdw map property temperature colorScheme @x. ||x = color(schemeName, min, max)| (Note: To get just the {{r g b}} color value of a named color, use the .color function rather than the function described here: print "red".color reports {{255.0 0.0 0.0}}.) Coloring by atom property and mapping of isosurfaces utilize mapped color schemes, which are sets of colors that are associated with given ranges of values. The color function returns information relating to these schemes. The basic form of the function, with one or three parameters returns an associatvie array describing a color scheme such as "rwb" or "roygb" that has been mapped from value min to value max with the keys listed below. If schemeName is "", then the current property scheme (default "roygb") is used.) The parameters min and max are optional and, if absent, will take the numbers 1 and N, where N is the number of colors in the color scheme if no property has been mapped, or the most recent range of values, if a property has been mapped.[|| "colors" | an array of points giving {{r g b}} on a 0 to 255 scale||"max" | the maximum value being mapped || "min" | the minimum value being mapped || "name" | the name of the color scheme || "reversed" | TRUE if min > max || "values" | an array of values associated with the two ends and each boundary between individual color of the scheme. (Note that if there are N colors, then there will be N + 1 values.) ||]
Thus, for example, print color("rwb", 0, 100)["colors"][1] prints the first color of the color scheme, and print color("rwb", 0, 100)["values"][0] prints the last value (max). ||x = color(schemeName, min, max, value)| Returns a color as {{r g b}} on a 0 to 255 scale from a specified color scheme that has been mapped using the specified minimum and maximum values. For example: print color("rwb", 0, 100, 30) returns {{255.0 144.0 144.0}}. ||x = color("toHSL",RGBcolor) | From an RGB color expressed as a point, {{100 50 100}} or color(100, 50, 100), or a hex name such as "[xFF00FF]", or a JavaScript color name such as "chartreuse", returns an {{h s v}} point with hue ranging from 0 to 360, saturation ranging from 0 to 100, and lightness ranging from 0 to 100. ||x = color("toRGB",colorNameOrHSLcolor) | From a color name or hex code such as [xFF00FF] or an HSL color expressed as a point, {{100 50 100}} or using color(100, 50, 100), returns an {{r g b}} point with red, green, and blue ranging from 0 to 255. (Note that you can also use colorName.color to get the color of a JavaScript color: print "red".color, for example.)|| x = color("$isosurfaceID") | Returns the color scheme associative array (see above) associated with this isosurface. || x = color("$isosurfaceID", value) | Returns the color as {{r g b}} associated with the given value of a mapped isosurface. ||x = compare({{atomset1}}, {{atomset2}})|Returns a 4x4 matrix that gives the optimal rotation (x%1) and translation (x%2) to superimpose the atoms in {{atomset1}} onto the atoms in {{atomset2}} using the closed-form quaternion method. One or the other or both of the atom sets may be replaced by an array of points. The number of atoms/points must be the same in each set, or "NaN" will be returned instead. ||x = compare({{atomset1}}, {{atomset2}}, "stddev")|Returns the standard deviation of atom positions for the optimal rotation and translation to superimpose the atoms in {{atomset1}} onto the atoms in {{atomset2}}. The number of atoms must be the same in each set, or "NaN" will be returned instead. || x = compare({{atomset1}}, {{atomset2}}, "ISOMER")| Carries out a comparisons of atoms in the two atom sets and returns one of the following strings : [|| CONSTITUTIONAL ISOMERS | having the same molecular formula but not the same connectivity || ENANTIOMERS | configurationally enantiomeric (mirror images) || DIASTERIOMERS | differing in one or more stereocenters (including double bonds) || CONFORMATIONAL ISOMERS | having the same constitution and configuration, but differing significantly in conformation (having root-mean-square deviations greater than ?? || IDENTICAL | the same, constitutionally, configurationally, and conformationally || NONE | no relationship ||] ||x = compare({{atomset1}}, {{atomset2}}, "SMARTS", SMARTSstring)|Uses a SMARTS (substructure) description to find the first instance of atoms in the first structure that correlate one-for-one with atoms in the second structure, then finds the rotation and translation that best aligns them. Returns a 4x4 matrix. ||x = compare({{atomset1}}, {{atomset2}}, "SMARTS", SMARTSstring, "stddev") | Same as above, but returns the standard deviation. ||x = compare({{atomset1}}, {{atomset2}}, SMARTSstring, "BONDS") |Carries out a SMARTS match (or SMILES if the string is "SMILES" itself) on the two atom sets. Returning an array of n*6 elements, where n is the number of dihedral angle matches. Each set of six elements indicates the four atoms involved in the dihedral, the angle in the first atom set, and the angle in the second atom set. If there is no match, the string "NaN" is returned. This function is run internally by Jmol when carrying out flexible fits using the {#.compare~} command and the BONDS option. ||x = compare({{atomset1}}, {{atomset2}}, "SMILES", SMILESstring) |Same as "SMARTS", but does a whole-molecule comparison. ||x = compare({{atomset1}}, {{atomset2}}, "SMILES", SMILESstring, "stddev") |Same as "SMARTS", but does a whole-molecule comparison. ||x = compare({{atomset1}}, {{atomset2}}, "MAP", SMILESstring)[1] |Reports a mapping of atoms in one model to the atoms in another model, using SMILES. The result is array of [a1 a2] correlations, ordered by atom position in the SMILES string. a1 and a2 are atomIndex values. (AtomIndex is zero-based, running from the first atom of the first-loaded model to the last atom of the last-loaded model.) ||x = connected(...{{atomSet}})|See the discussion of connected() at {#.atomexpressions~atom expressions}.||x =connected(...{{atomset1}}, {{atomset2}})| Specifying two atom sets in a CONNECTED function returns the bond set [{{...}}] including all such bonds. This bond set can be selected, colored, sized using {#.wireframe~}, and hidden or displayed. || connected({{bondSet}},...)|returns a subset of the specified bonds that match additional criteria, for example: x = connected({{*}}.bonds, "DOUBLE");connect @x radius 0.1 or x = connected({{*}}.bonds, 1.3, 1.5);color @x blue.|| x = contact({{atomset1}}, {{atomset2}})
x = contact(nPercent, {{atomset1}}, {{atomset2}})
x = contact(distance, {{atomset1}}, {{atomset2}})|returns the atoms in {{atomset2}} within van der Waals contact of {{atomset1}}. {{atomset2}} is optional and defaults to "not atomset1". Optional parameters include a percent van der Waals radius expressed as an integer or a distance in Angstroms do be added to the van der Waals radius, expressed as a decimal number ||x = cos(y)|the cosine of y, where y is in degrees ||x = cross(a,b)| the cross product of two vectors or points.||x = data({{atomset}},type)|creates model file data of the type "MOL", "PDB", or "XYZ" for the selected atom set. Starting with Jmol 14.4, if type is lower case "xyz", then only the atom data lines are written, not the header lines of the XYZ file format.||x = data({{atomset}},type,TRUE)|Adding a third parameter TRUE specifies that all trajectories should be returned.||x = data("dataset_name")|places the text of the data set created using the {#.data~} command into variable x.||x = data(stringData,fieldOrColumn,columnCount,firstLine)|separates stringData into lines, then reads lines starting from firstLine (1 being the first line). Data are read from free-format field or column fieldOrColumn. If the data are free-format, then set columnCount = 0, otherwise columnCount indicates the number of columns to read for each data point. The data() function returns a newline-separated list, which can be read directly into atomic properties, for example, using {{*}}.partialCharge = data(load("mydata.mol2"),9,0,7).||x = distance(a,b)| The distance from the geometric center of a to the geometric center of b, where a and b are atom expressions, coordinates, or planes.||x = dot(a,b)| the dot product of two vectors (or points). ||x = eval(expr)|indirect evaluation of a Jmol math expression. ||x = eval("JSON",json) | evaluate a JSON string to give an associative or serial array; for the reverse, see format("JSON",data) ||x = file("filename")| the full path to the indicated file name. Note that file("?") displays a file dialog, which allows the user to navigate to a different directory, and file("") returns the full path to the current default directory. ||x = format("ARRAY",data)|Makes a copy of the data in array form, turning string and byteArrays into integer arrays based on characters or bytes, respectively.||x = format("BYTEARRAY",data)|Copies the data into a byteArray. Data may be an array, a byteArray, a string, or a base64-encoded array.||x = format("BASE64",data)|Generates base64 code for data contained in a variable, prepending the string with ";base64,".||x = format("JSON",data)|Generates JavaScript Object Notation (JSON) code for data contained in a variable; for the reverse, see eval("JSON",json).||x = format(sprintfFormat, a, b, c, ...)|This function creates a string using a format similar to that used in C++ to format a set of variables into a string with special codes that start with %. While not an exact implementation of this format, there are strong similarities. Here a, b, and c are variable names, and sprintfFormat is the format string containing a %n.mX code for each variable. As for {#.labelcolumnformattingandscientificnotation~labels}, n and m indicate column formatting and precisions. Both n and .m are optional. Here, X indicates the variable type , whereas in, for example, {{*}}.label("%a"), X represents an atom property, and the type of formatting is determined automatically from that. The options for X include:
[|| i or d| integer. Either i or d can be used synonymously.|| f|float/decimal. A negative value for m indicates to use scientific notation with a total of -m digits. The default is full width, full precision.|| e|exponential (scientific notation). "%8.3e" is equivalent to "%8.-3f". || p|point. Each of the three coordinates is formatted according to the specified width and precision. The default is %6.2p. || q|quaternion/plane/axisangle. Each of the four elements of the vector {{x y z w}} are formatted according to the specified width and precision. The default is %6.2q|| s|string. Values for precision, m, determine maximum number of characters starting from the left (m > 0) or right (m < 0). So, for example, print format("%0.-3s","testing") prints "ing". ||]
For example: calculate straightness;print format("average straightness = %4.2f", {{*}}.straightness)||x = getProperty(infotype,parameters...)| The {#.getProperty~property} of the given infotype is returned as a Jmol math list; print getProperty() by itself giving the list of available types. Each property type has its own intrinsic structure, but in general the parameters may include an initial atom set specification followed by one or more key values and, in the case of arrays, an item selector. For example,
print getProperty("boundboxInfo","center")
x = getProperty("atomInfo",{{atomno=3}})
x = getproperty("bondInfo",{{*}},2,"atom1", "sym")
||x = getProperty("JSON",infotype,parameters...)| An optional format "JSON" may be included in the getProperty function as the first parameter to indicate that the data should be converted to JSON format. The JSmol call from JavaScript Jmol.evaluateVar(app,expr) uses the getProperty method to evaluate an expression and return its value in JavaScript format, via JSON.||x = getProperty(array,query)| Use {#.JmolSQL~} to extract information from a serial or associative array. || x = hkl(a,b,c)| generates the plane associated with a given set of Miller plane indices. || x = intersection(plane1, plane2)|returns the line of intersection between two planes in the form of an array: [point,vector] or "" if the planes are parallel. ||x = intersection(point,plane)|returns the nearest point on a plane to a given point. ||x = intersection(point,vector,plane) |returns the point of intersection of a plane with a line trough the given point with in the direction of the given vector. ||x = intersection(point1, vector1, point2)|returns the nearest point to point2 on the line through point1 along the vector vector1.||x = intersection(point1, vector1, point2, radius2)|returns an array of 0 to 2 points of intersection of a line through point1 along vector1 with a sphere of radius radius2 centered on point2 ||x = javascript("...")|returns the result of evaluating the specified JavaScript. Applet only; disallowed if the applet has been started with Info.allowJavaScript = false.||x = label(...)| See x = format(...), above.||x = load("filename")| Load the data from the specified file into variable x.||x = load("filename", nBytesMax)| Load the data from the specified file into variable x, but no more than the specified number of bytes. Since this function returns "java.io.FileNotFoundException: ..." when working from a local drive, the function x = load("filename", 0) can be used to test for the existance of a file. If the function returns the empty string "", then the file exists; if it returns an error message, then the file does not exist. So, for example, the following code loads a PDB file from the RCSB only if not found on the local drive:
if (load(pdbid + ".pdb", 0) == "") {{load @{{pdbid + ".pdb"}} }} else {{load @{{"=" + pdbid}} }}
|| x = load("filename",TRUE) |Adding a second parameter TRUE creates a binary associative array variable x that contains the contents of the file. Data are stored as raw bytes (but will appear as a string in the PRINT command). If the file is a standard file, the key "_DATA_" will hold the file data. If the file is a PNGJ file, the key "_IMAGE_" will hold the image data and additional files will be keyed by file name. If the file is a zip file, the files will be keyed by file name, but no "_IMAGE_" key will be present (presumably). Note that x = load("myfile") without TRUE loads just the file list for PNGJ or ZIP files, not the actual data.|| x = measure(...)| The measure function requires from two to four atom expressions and/or points in space and returns associated measurements in a string, one measurement per line. Additional optional parameters include minimum and maximum ranges, the designations "connected" or "notConnected", the units "nm", "nanometers", "pm", "picometers", "angstroms", "ang", or "au", and a format string in quotes similar to that used by the {#.measure~} command. ||x = now()| returns the time in milliseconds since some old date; can be used for timing scripts.||x = now(n)|(n integer) returns the number of milliseconds since time n: x = now();.......;print now(x);. || x = now(msg)|(String msg) returns the date in the Java SimpleDate format "EEE, d MMM yyyy HH:mm:ss Z" followed by tab and the message. || x = now(msg, format)|(String msg) returns the date in the specified Java SimpleDate format followed by a tab and the message. Special formats include "iso8601" ("yyyy-MM-dd\'T\'HH:mm:ss") and "iso8824" ("D:YYYYMMddHHmmssX\'00\'"). Note: With JSmol prior to Jmol 15, if the format is provided, it must be one of these two special formats.|| x = plane(pt1, pt2)| the plane that bisects the line connecting pt1 and pt2. ||x = plane(pt1, pt2, f)| the plane perpendicular to the line connecting pt1 and pt2 and containing the point that is fraction f of the way from pt1 to pt2. ||x = plane(pta,ptb,ptc,ptd)|creates an {{x y z w}} plane from the first three points, and assigns the signs of x, y, z, and w to correspond to a positive distance to ptd as measured with the distance() function. Parameters may be mathematical expressions. ||x = plane(a, b, c, d)|creates the four-vector {{a b c d}}, which represents a plane satisfying the equation ax + by + cz + d = 0||x = plane("{{a b c d}}")|creates an plane satisfying the equation ax + by + cz + d = 0 from a string equivalent. As for all Jmol math expressions, parameters may be mathematical expressions. ||x = plane(pta,ptb,ptc)|creates an {{x y z w}} plane through the three given points, which may themselves be mathematical expressions that evaluate to {{x y z}} points or atom expressions. || x = plane(r,theta,phi)|generates a plane that is perpendicular to the vector from {{0 0 0}} to the point defined by the given r, theta, and phi. The vector is generated by sequential rotation of a vector along the Z axis first by phi around the X axis, then by theta around the Z axis: v = R[z,theta]R[x,phi]{{0 0 r}}. ||x = point(a,b,c)|Creates an {{x y z}} point. Parameters may be mathematical expressions.||x = point({{x,y,z}}, true)|gives the screen coordinates {{sx, sy, sz}} corresponding to the 3D coordinates {{x, y, z}}. ||x = point({{sx,sy,sz}}, false)|gives the 3D coordinates {{x, y, z}} cooresponding to the screen coordinates {{sx, sy, sz}}.||x = point("{{x,y,z}}")|creates an {{x y z}} point from the string equivalent. Parameters may be mathematical expressions. ||x = point(x)|Rounds x down (toward 0) to the nearest integer. Note that x%0 rounds to the NEAREST integer value, instead.||x = point(unitcell, {{i, j, k}})|returns the Cartesian point corresponding to a specific coordinate in a unit cell system. unitcell is an array of the form [origin, va, vb, vc]. {{i j k}} is a point in fractional coordinates. Does not require actual setting of the model\'s unit cell||x = pointgroup([array of points],center)|Analyzes the point group of a set of points; center is optional and defaults to the geometric center of the array of points. Creates a map with and extensive list of keys, including (if appropriate): C2, C3, Ci, Cs, S6, detail, distanceTolerance, linearTolerance, nAtoms, nC2, nC3, nCi, nCn, nCs, nS6, nSn, nTotal, name, points, principalAxis, and principalPlane.||x = pointgroup("spacegroup",@1)|Analyzes the point group (crystal class) of a crystal using three irrational coordinate points to generate all possible operators. Creates a map with and extensive list of keys, including (if appropriate): C2, C3, Ci, Cs, S6, detail, distanceTolerance, linearTolerance, nAtoms, nC2, nC3, nCi, nCn, nCs, nS6, nSn, nTotal, name, points, and principalAxis, and principalPlane.||x = polyhedra(); x = polyhedra(n)| the atom centers for all polyhedra or polyhedra with a specific number of vertices, respectively. ||x = polyhedra(smilesString); x = polyhedra(smartsString)| These two functions can be used search for matching polyhedra (atom environments), either topologically (using a SMARTS string; only *) or including atom identity (SMILES string; with atom names). An atom set is returned. Note that the SMILES and SMARTS strings created by Jmol in association with {#.polyhedra~} describe the connectivity of the faces and do not include the central atom itself. ||x = prompt(message)|displays a pop-up message box and waits for the user to press OK||x = prompt(message,defaultInput)|displays an input dialog allowing the user to enter some text and press OK (or ESCAPE to cancel). If canceled, returns "null". ||x = prompt(message,buttonText,TRUE)|displays a message box with buttons and returns the label of the button that was pressed. The parameter is a list of button labels separated by ¦, for example: "Yes¦No" or "OK¦cancel" or "Spacefill¦Wireframe¦Ball&Stick".||x = prompt(message,buttonArray)|displays a message box with buttons based on the values in the array parameter and returns an integer indicating which button was pressed (starting with 1, or -1 if ESCAPE was pressed). ||x = quaternion()| The current model rotation in the form of a quaternion. Same as x = quaternion(script("show rotation")) ||x = quaternion("best")| The {#.rotate_best~"best"} model rotation in the form of a quaternion. Same as x = quaternion(script("show best rotation")) ||x = quaternion({{x y z}},theta)|the quaternion {{x y z w}} associated with a rotation of theta degrees (counter-clockwise) around axis {{x y z}}. {{x y z}} need not be a unit vector -- Jmol will normalize it automatically. ||x = quaternion("{{x y z w}}")|the unit quaternion {{x y z w}} produced by normalizing the specified quaternion. That is, where theta is the rotation angle, and q0 = w = cos(theta/2), {{x y z}} = sin(theta/2) * unitNormal. ||x = quaternion(q0, q1, q2, q3)|the unit quaternion {{x y z w}} produced by normalizing the specified quaternion. Note that q0 is first in the list of parameters, even though Jmol will store the quaternion in the form {{q1/f, q2/f, q3/f, q0/f}} where f is sqrt(q0*q0 + q1*q1 + q2*q2 + q3*q3). ||x = quaternion({{m00 m10 m20}}, {{m01 m11 m21}})|the unit quaternion corresponding to the rotation matrix having the first two column vectors indicated. ||x = quaternion({{center}},{{x-axis point}}, {{xy-plane point}})|the unit quaternion associated with a frame that has a center at the first parameter position, an x axis in the direction of the second parameter position, and a y axis in the plane of the three parameter positions. These parameters may be atom expressions. For example, x = quaternion({{215.CA}},{{215.C}},{{215.N}}) creates the "standard" quaternion for residue 215. The z axis will be generated using the x and y axes and the right-hand rule. ||x = quaternion({{atom}})| the quaternion associated with the an atom (or the first atom in the atom set) based on the setting of set quaternionFrame.||x = quaternion({{atomset}}, nMax)| an array of quaternions, one per residue, up to nMax long (or all if nMax is <=0). Quaternions are created based on the setting of set quaternionFrame.||x = quaternion({{atom1}}, {{atom2}})| the quaternion difference of the two residues containing the specified atoms (or the first atom in each set, if applicable), created based on the setting of set quaternionFrame. An optional last parameter "relative" utilizes quaternion left-division. The quaternion relative difference represents the necessary rotation to get from q1 to q2 within the reference frame of q2 rather than the standard reference frame ||x = quaternion({{atomset1}}, {{atomset2}}, nMax)| an array up to nMax long (or all if nMax is <=0) of quaternion differences, one per residue pair in the two sets, created based on the setting of {#set(misc)~set quaternionFrame}. An optional last parameter "relative" utilizes quaternion left-division. ||x = quaternion(quaternionArray)| calculates the spherical mean (see {http://math.ucsd.edu/%7Esbuss/ResearchWeb/spheremean/~Buss and Fillmore, 2001}) of the array of quaternions.||x = quaternion(quaternionArray, true)| returns the standard deviation for the calculated spherical mean (see {http://math.ucsd.edu/%7Esbuss/ResearchWeb/spheremean/~Buss and Fillmore, 2001}) of the array of quaternions.||x = quaternion(quaternionArray1, quaternionArray2)| an array of quaternion differences of the two array elements, taken a pair at a time. An optional last parameter "relative" utilizes quaternion left-division. ||x = random()
x = random(n)
x = random(m,n)
x = random(m,n,seed)|Generate a random number in the range [0,1), [0, n), or [m, n). Optionally set the seed (resetting the generator to a predefined point).||x = script("...")|returns the output from the specified script command; particularly informative script commands include {#.getProperty~} and {#.show~}.||x = script("...", appletName)|returns the output from the specified script command run in one or more applets. For example, print script("show orientation moveto", 2) will print the orientation moveto command for an applet with name "2" or, if that does not exist, "jmolApplet2". See {#.script~} for details. ||x = search(smilesString)| find atoms matching the given smiles string, which may include bond types such as = or - between atoms. More powerful SMILES/SMARTS functions include {#.jmolsmarts/smiles~find() and search()}. || x = select(x;{{a}};b)|selects atoms from the atom expression {{a}} based on the boolean expression b. Note the use of semicolons, not commas, to separate the three components of this function. The variable x is local to the function, and when it appears in the boolean expression in the form x.property represents a single atom of the atom expression. For example, x = select(a;{{*}};a.distance({{0 0 0}}) > 3 and a.elemno > 18). select() functions can be nested -- just use two different variable names: x = select(x;{{*.ca}};x.phi < select(y; {{*.ca}}; y.resno = x.resno + 1).phi). The select() function provides a powerful selection mechanism that can utilize any Jmol math expression involving properties of an atom. (In contrast, {#.select~} command comparisons are limited to =, <, >, <=, and >=, and values are rounded to the nearest 1/100th). ||x = show(y)|returns the result of the {#.show~} command; the same as x = script(\'show y\') but simpler to implement.||x = sin(y)|the sine of y, where y is in degrees ||x = sqrt(y)|the square root of y ||x = symop(...)|same as {{thismodel}}.symop(...)||x = unitcell()| returns a "unit cell" array containing [origin, va, vb, vc] suitable for use in the command UNITCELL @x.||x = unitcell(uc)| returns a copy of a unit cell.||x = unitcell(uc, "reciprocal")| returns the reciprocal lattice for specified unit cell.||x = unitcell("reciprocal")| returns the reciprocal lattice for current model\'s unit cell.||x = unitcell("primitive")| returns the primitive unit cell for the current model. (Assumes current unit cell is conventional.)||x = unitcell("primitive", centering)| returns the primitive unit cell for the current model using the specified centering, which may be one of "P", "A", "B", "C", "I", "F", "R", "BCC", or "FCC". (Assumes current unit cell is conventional.)||x = unitcell("conventional")| returns the conventional unit cell for the current model. (Assumes current unit cell is primitive.)||x = unitcell("conventional", centering)| returns the primitive unit cell for the current model using the specified centering. (Assumes current unit cell is primitive.)||x = unitcell(ucconv, "primitive",centering)| converts conventional to primitive based on a centering.||x = unitcell(ucconv, "conventional",centering)| converts primitive to conventional based on a centering.||x = unitcell("a=10, b=10, c=20, alpha=90, beta=90, gamma=129")| returns a unit cell specified by a string.||x = unitcell(origin, [va, vb, vc])| returns a user-defined unit cell; just the array [origin, va, vb, vc].||x = unitcell(origin, pta, ptb, ptc)| returns a user-defined unit cell as [origin, pta - origin, ptb - origin, ptc - origin].||x = within(...)|returns a matching atom set for a wide variety of atoms that are in some way "within" another atom set or plane or "within some distance" of any one of a set of atoms or a plane.||x = within("SMARTS", smartsString,{{searchSet}})|returns any array of subsets of the atoms that match the given smarts string. If provided, only atoms in {{searchSet}} will be checked. More powerful SMILES/SMARTS functions include {#.jmolsmarts/smiles~find() and search()}. ||x = within("SMILES", smilesString,{{searchSet}})|returns any array of atom sets that match the given smiles string. If provided, only atoms in {{searchSet}} will be checked. More powerful SMILES/SMARTS functions include {#.jmolsmarts/smiles~find() and search()}. ||x = within(distance, point, [arrayOfPoints]) | Returns a subarray of points (not necessarily in the same order) that are within a given distance of a given point. ||x = within(tolerance, [arrayOfPoints]) | Returns a subarray of points that have no points within tolerance Angstroms of each other.||x = within(distance, TRUE, "UNITCELL", {{atomset}}) | This function iterates over all the atoms that could be loaded using crystallographic symmetry even if they have not been created. It returns an associative array with two keys: "atoms" and "points", each of which is an array. For example, x = within(4, true, "unitcell", {{selected}}).The atoms array lists atom indices for the equivalent originating atoms present in the model; the points array lists the coordinates of the atoms. The following DRAW command visualizes these points: draw width 0.2 points @{{x["points"]}} color red mesh nofill.||x = write(...)|the output of the {#.write~} command is loaded into variable x. The parameters are those of the write command. For example, x = write("PDB") or x = write(quaternion, "r", "difference2"). Note that x = write("PNGJ") creates a binary associative array for which the keys are file names and the values are their associated file data, equivalent to writing a PNGJ file. The image for this PNGJ file will be the value associated with the "_IMAGE_" key. Likewise, write("ZIP") and write("JMOL") create ZIP files that can be loaded back into Jmol using the LOAD command. The difference between the ZIP and JMOL options is that the JMOL option stores all files necessary to recreate the visualization regardless of source, whereas ZIP saves only local files, leaving only references to remote resources involving http: or https:. Note that since more key/value pairs can be added to x using x.push(key,value) or can be removed using x.pop(key), this mechanism allows one to add (or remove) files from a PNGJ, ZIP, or JMOL collection. A subsequent WRITE VAR x ... command can then be used to save the modified PNGJ, ZIP, or JMOL file to disk. ||]','TEXT','x = f(y) functions','') newCmd('','11.7.37','','*v+11.2;','These functions operate on coordinates, either the geometric center of a set of atoms expressed as {{atom expression}} or a coordinate point expressed as {{x y z}}. The unit cell system used is the currently defined {#.unitcell~} for the current model. If more than one model is visible, all coordinates are considered Cartesian coordinates, and these functions are not distinguishable.
[||x = pt.xyz | the Cartesian coordinates for the point. For example, if pt = {{1/2 3/2 1}} in an orthonormal unit cell with a = 28.0, b = 5.04, c = 6.04, then pt.xyz would equal {{14.0 7.56 6.04}}. || x = pt.fxyz| the fractional coordinates of a point. In the same case, pt.fxyz would be {{0.5 1.5 1.0}}. ||pt.fx, pt.fy, pt.fz| The fractional x, y, and z coordinates, respectively.|| x = pt.sxyz| the screen pixel coordinates of the point in space. A pt.sxyz of {{0 0 1000}} is in the bottom left corner, with a z-value of 1000. All z-value are ≥ 1, with 1 indicating "clipped" because the point is too close to the user. These values depend upon screen size and the sort of perspective model being used. ||pt.sx, pt.sy, pt.sz| The screen x, y, and z coordinates, respectively. || x = pt.uxyz| the unit-cell normalized point in the range [0,1). In this case, pt.uxyz would be {{0.5 0.5 0}}. ||pt.ux, pt.uy, pt.uz| The unit cell x, y, and z coordinates, respectively. || pt.x, pt.y, pt.z| The x, y, or z component of the point, regardless of the unit system. ||]
Note that the fractional notation {{1 1/2 1}} is immediately converted to Cartesian coordinates, so {{1/2 3/2 1}} = {{14.0 7.56 6.04}} in this case, and {{1/2 3/2 1}}.y = 7.56, whereas {{1/2 3/2 1}}.fy = 1.5, and {{1/2 3/2 1}}.uy = 0.5. Note that if vibration vectors are set using, for example, {{atomno=3}}.vxyz = {{1/2 1/2 1/2}}, then the frame command should be sent so as to update the popup menu so that the vibration menu item is enabled .','TEXT','coordinate transforming .xxx functions','') newCmd('','','','*v+11.8;','These modifiers can be used with a number of different variable types.
[||x = y.array|Forces y into an array format. If y is already an array just returns that; otherwise returns an array created from y. Variables of type matrix3f and matrix4f are converted to standard 3x3 or 4x4 arrays; others are returned as [ xxx ]. This function can be used to force an array where a string or other single value might be returned. In this way it is similar to y.all, but whereas y.all only works on atom properties, y.array works on any type of value. ||
[||.all| If a property, such as {{selected}}.vanderwaals, then appending .all creates a list of those measures. This list can be used to transfer one property to another, as in: {{*}}.partialCharge = {{*}}.temperature.all, which would allow temperature data to be used for partial charges in an isosurface molecular map MEP command, for instance. Note that the "list" is really an array of string values. ||.average|the average value (the default modifier). ||.max|the maximum value, for example: {{*}}.temperature.max||.min|the minimum value, for example: {{*}}.partialCharge.min ||.pivot| Creates a pivot table from an array . For example:
print {{*}}.element.all.pivot
{{:> "Ag" : 1561:> "Cd" : 1360
}}
load $caffeine
print {{_N}}.bondCount({{_C}}).pivot
{{:> "2" : 1:> "3" : 3
}}
||.reverse|reverses an array IN PLACE (does not make a copy). Thus, the preferred command is simply xxx.reverse.||.sort|sorts an array IN PLACE (does not make a copy). Thus, the preferred command is simply xxx.sort. An optional parameter allows for sorting a multidimensional array based on a specific "column" of data: For example, x = [ [1,"orange"], [2,"apple"], [3, "banana"] ]; x.sort(2) sorts this array to read: [[2,"apple"], [3, "banana"], [1,"orange"] ] ||.stddev|the standard deviation, for example: print {{helix}}.straightness.stddev ||.sum|the sum of the values ||.sum2 |sum of squares ||]','TEXT','[array].xxx or .propertyName.xxx modifiers','') newCmd('','','','*v+11.8;','These functions operate on each atom in an atom expression. In addition to these are all of the atom properties described under the heading {#.atomexpressions~atom expressions}, in which case they give the average value. For example: x = {{oxgyen}}.temperature. Several more functions that act on atom expressions are in the {#.functionsx=fyfunctions~section below}. [||x = y.atoms|The atoms associated with a set of bonds.||x = y.bonds| The bonds associated with the specified atoms, using the current setting of bondModeOr (true, bonds having one OR the the other atom within this set; or false, bonds having BOTH atoms within the set) ||x = y.boundbox|A Jmol math list containing the boundbox center, vector to corner, and two opposite corners associated with this atom set ||x = y.color|The average color of the atoms in the set y expressed as a {{r,g,b}} coordinate in color space. If y is already a point, then .color converts this to a string of the form [[xRRGGBB]]. See also {#.functionsgeneralxxxfunctions~general x = y.color}, above. || x = y.ident| a list of the standard identity labels for the elements of y, either atoms or bonds.||x = y.length|In the case of x a set of bonds, the average length of the bonds. In all other cases, the length of the data. For example: x = {{carbon}}.bonds.length, x = {{*}}.length. To check the size of a bondset, use .size. ||x = y.size|The number of selected atoms or bonds in a bitset. ||]','TEXT','{atomExpression}.propertyName','') newCmd('','','','*v+11.2;','These functions operate on the individual elements of some group or listing, returning a modified listing.[||x = data1.add(data2)|Specifically for use with data set lists, adds each element of data1 to its corresponding element in data2 and returns the result. If data2 is a simple number, adds that number to each element of data1.||x = data1.add("\\t",data2)|Specifically for use with data set lists, creates a new column separated from the previous with a tab. ("\\t" may be replaced with whatever separation one desires. ||x = mapOfMaps.array(key)|This powerful method creates an array from a map of maps, moving the top-level keys of mapOfMaps to values of the specified key. Thus, starting with
x = {{ :>"a" : :>{{ :>>"aa" : 1 :>>"ab" : 2 :>}} :>"b" : :>{{ :>>"ba" : 3 :>>"bb" : 4 :>}} :>"c" : :>{{ :>>"ca" : 5 :>>"cb" : 6 :>}}
}}
x.array("id") gives:
:>{{ :>>"aa" : 1 :>>"ab" : 2 :>>"id" : "a" :>}} :>{{ :>>"ba" : 3 :>>"bb" : 4 :>>"id" : "b" :>}} :>{{ :>>"ca" : 5 :>>"cb" : 6 :>>"id" : "c" :>}}
For this function to be successful, all values in mapOfMaps must be maps themselves. ||arrayOfMaps.array(key)| Carries out the reverse process of mapOfMaps.array(key), placing the elements of arrayOfMaps into a map, such that the top-level key in the map is the value of the given key. The key must be found in each element map of the array.||x = array.bin(low,high,binSize,asArray)| Operating on an array, delivers an array that counts the number of values in specific bins defined by a low limit, a high limit, and a bin size. If the fourth parameter is TRUE, then creates an array of arrays for which the first value is the lower limit and second value is the count. Values equal to the lower limit of a bin are included in that bin. Values less than the low limit or higher than or equal to the upper limit are ignored (starting in Jmol 14.4.4_2016.04.04). ||x = array.bin(low,high,binSize,fieldName, asArray)| For an array of associative arrays, creates a pivot table of bins from one of the associative array\'s values. Also adds keys "_bin", "_binMin", and "_binMax" to the initial arrays. For example, if x = [{{"energy":2.345,"model":1}},{{"energy":1.5,"model":2}},{{"energy":2.36,"model":3}}], then x.bin(1,3,0.5,"energy") will be [ 0,1,2,0 ].. Similarly for sequential arrays, adding TRUE for the fourth parameter, creates an array of arrays. ||x = {{atomset1}}.bondCount({{atomset2}}) |Counts bonds to a specific set of atoms. ||x = arrayOfArrays.col(n)| selects out the nth column from an array of arrays. For example, retrieving the third column from CSV data: col3 = load("data.csv").split("",true).col(3).||x = array.count()| Operating on an array, .count() returns an array of [value, count] pairs. Thus, for example, {{*}}.radius.all.count() would return a list of all the radii of all the atoms and how many of each radius value there are. || x = y.count(z)| returns how many z there are in y, which should be either an array or a string.||x = a.cross(b)| the cross product of two vectors or points.|| x = y.distance(z)|The evaluation of this function depends upon the type of variables y and z:[||y type|z type|evaluation|| atomset| atomset| The average distance from atoms of y to the geometric center of z; same as y.distance(z.xyz).|| atomset| point | The average distance from atoms of y to the point z.|| point| atomset | The distance from the point y to the geometric center of z.|| point| plane | The distance from point y to plane z. The absolute value of the result depends upon the winding of the points used to create the plane, using a right-hand winding rule.||]|| x = y.distance(plane, point)|The distance of y to a plane, using a point for a reference, giving a positive value if the y and the point are on the same side of the plane and a negative number if they are on opposite sides of the plane. ||x = {{atomset1}}.distance.all({{atomset2}}| Generates an array of arrays of distances indicating distances from every atom in {{atomset1}} to every atom in {{atomset2}}. ||x = {{atomset1}}.distance.min({{atomset2}}, asAtomSet) | If asAtomSet is true, returns the closest atom in atomset1 to any atom of atomset2; if false or omitted, returns an array listing the distance of each atom in atomset1 to the closest atom in atomset2. This array can be used to assign properties to atomset1: {{1.1}}.property_d = {{1.1}}.distance.min({{2.1}}); color {{1.1}} property_d. ||x = {{atomset1}}.distance.min({{point}}, asAtomSet) | If asAtomSet is true, returns the atom in atomset1 closest to the specified point;if false or omitted, returns the closest distance to the specified point from any atom in atomset1.||x = {{atomset1}}.distance.min({{atomset2}}).min | returns the shortest distance from any atom in atomset1 to any atom in atomset2.||x = {{atomset1}}.distance.max({{atomset2}}, asAtomSet) | If asAtomSet is true, returns the furthest atom in atomset1 to any atom of atomset2; if false or omitted, returns an array listing the distance of each atom in atomset1 to the furthest atom in atomset2.||x = {{atomset1}}.distance.max({{point}}, asAtomSet) | If asAtomSet is true, returns the atom in atomset1 furthest from the specified point;if false or omitted, returns the furthest distance to the specified point from any atom in atomset1.||x = {{atomset1}}.distance.max({{atomset2}}).max | returns the furthest distance from any atom in atomset1 to any atom in atomset2.||x = data1.div(data2)|See data1.add(). Division by 0 results in the value "NaN", meaning "not-a-number".||x = a.dot(b)| the dot product of two vectors or two points or a point with a plane (as its normal). ||x = y.find("s")|finds the first location of "s" in y or, in the case of y being a set of lines, selects only the lines of y containing "s".||x = y.find("pattern","flags")| Searches string or list y for a {http:
[|| |(no flags) Returns the position in the string containing the pattern, starting with 1, or 0 if the pattern is not found. For lists, returns a sublist containing the elements that match.||i |(case-insensitive) Match upper or lower case letters.||v |(reverse match) With a string, v returns "false" if the string contains the match or "true" if it does not; with lists, v returns all elements of the list that do not contain the match.|| m|(return match) The m option allows returning only the portion of the string that matches (or, with vm, only the portion of the string that does NOT match). With lists, both m and vm return only elements that contain the match, but, as for strings, each element is returned as just the matching phrase or the reverse. ||]
Note that special characters such as \\S and \\d must be escaped with two back-slashes, and if they are introduced via JavaScript, they will need double escaping (four back-slashes). Examples include:
[||"this test is a Test".find("Test","") |16||print "this test is a Test".find("Test","i")|6||print "this test is a Test".find("Test","m")|Test||print "this test is a Test".find(" a test","v")|true (because it was not found)||print "this test is a Test".find(" Test","ivm")|this is a Test||print "this test is a Test".find("\\\\stest","m")| test||print "this test is a Test".find("\\\\stest","vm")|this is a Test||print script("show spacegroup all").split()
.find("Hall symbol:")|Hall symbol: P 1
primitive Hall symbol: P 1
Hall symbol: -P 1
primitive Hall symbol: P 1 -1
Hall symbol: P 2y
primitive Hall symbol: P 2y
...||print script("show spacegroup all").split()
.find("Hall symbol:").find("primitive","v")|Hall symbol: P 1
Hall symbol: -P 1
Hall symbol: P 2y
...||print script("show spacegroup all").split()
.find("Hall symbol:").find("primitive","v")
.find("Hall symbol:","vm")[1][3]| P 1
-P 1
P 2y
||] || {{CELL=nnn}}.find("CELLFORMULA") | calculates cell formula for specified unit cell, weighing interior 1, face 1/2, edge 1/4, vertex 1/8; selection should be a single packed unit cell; fails with "?" if end result is not integral. For example, load quartz.cif packed;print {{*}}.find("cellFormula") gives "O 6 Si 3".|| {{CELL=nnn}}.find("CELLFORMULA", TRUE) | calculates the empirical cell formula for specified unit cell, for example: load quartz.cif packed;print {{*}}.find("cellFormula", TRUE) gives "O 2 Si 1".||x = {{atomExpression}}.find("chemical",type) | Determines or looks up the specific type of information for this subset of atoms. type may be one of: "smiles", "inchi", "stdinchi", "inchikey", or "stdinchikey". In the case of InChIs and InChIKeys, the return does not include the prefix "InChIKey=" or "InChI=". ||x.find("crystalClass", pt) | Generates a list of points based on a model\'s crystal class (point group). Uses x to find the unit cell and space group; pt is an optional generator, which defaults to x[1]. For example:
:>load =ams/calcite 1 :>x = {{*}}.find("crystalClass",@3) :>draw points @x :>polyhedra ID p {{0 0 0}} to @x
||x = {{atomExpression}}.find("smartsString", "BONDS") | Finds the first match in the structure to the given SMARTS string and returns an array of all the dihedrals involving the matching atoms. Each dihedral is expressed as an ordered list of the four atom indices. ||x = {{atomExpression}}.find("MF")| finds the molecular formula of the atom expression.||x = {{atomExpression}}.find("MF",formula)|finds the subset of atoms in this atom expression that matches the given formula, for example: {{xxx}}.find("MF","CH2O").||x = {{atomExpression}}.find("MF",TRUE)|finds the empirical formula for the specified atoms. For example, load $glucose; print {{*}}.find("MF",true) displays "H 2 C 1 O 1".||x = {{atomExpression}}.find("SEQUENCE")| returns the Jmol SMARTS/bioSMILES sequence for the specified atoms. An optional second parameter TRUE adds crosslinking.||x = smilesString2.find("SMILES",smilesString1, exactMap, allMappings) | Checks to see if a structure created from smilesString1 would match the structure associated with smilesString2, thus allowing Jmol to match two SMILES strings without need of "canonicalization." Boolean parameters exactMap and allMappings determine the value returned: [|||exactMap|allMappings|return value|||false|true| (default) Returns -1 if a SMILES parsing error is found, or the number of atoms in smilesString2 matched. For example, "O[C@](F)(Cl)I".find("SMILES","[C@](O)(F)(Cl)I", false, true) would return the value 5, meaning all 5 atoms of O[C@](F)(Cl)I were a match. A return value of 0 means the pattern was not found; a return of -1 indicates there was a problem parsing one or the other of the SMILES strings. |||false|false| 1 if a SMILES parsing error is found, 0 if no structure is found, or 1 if any match is made.|||true|true| an array of integer arrays listing all possible mappings of atoms in smilesString1 onto smilesString2.|||true|false| an integer array of the first match of atoms in smilesString2. Numbers refer to positions along smilesString1. So, for example, "CCCO".find("SMILES", "OCCC", true, false) evaluates to [3,2,1,0].||]||x = smilesString.find("SMARTS",pattern, exactMap, allMappings) | Checks to see if a substructure created from pattern would be found in the structure associated with smilesString. Boolean parameters exactMap and allMappings determine the value returned: [|||exactMap|allMappings|return value|||false|true| (default) -1 if a SMILES parsing error is found, empty array if no substructure is found, or the array of all atoms in smilesString matched by any possible mapping of the atoms in pattern. So, for example, "CCCC".find("SMARTS","CC",false,true) evaluates to [0, 1, 2, 3].|||false|false| -1 if a SMILES parsing error is found, 0 if no substructure is found, or 1 if any substructure is made.|||true|false| an integer array of the first match of atoms in smilesString. Numbers refer to positions along smilesString. So, for example, "CCCO".find("SMARTS","OCC",true,false) evaluates to [3,2,1].|||true|true| an array of integer arrays listing all possible mappings of atoms in pattern into smilesString.||]||x = smilesString.find("chemical",type) | Determines or looks up the specific type of information for this SMILES string. type may be one of: "smiles", "inchi", "stdinchi", "inchikey", or "stdinchikey". In the case of InChIs and InChIKeys, the return does not include the prefix "InChIKey=" or "InChI=".||x = smilesString.find("SMILES","MF")| returns the canonical molecular formula for smilesString, including all implicit H atoms.||x = smilesString.find("SMARTS", "MF")|returns the canonical MF for the smilesString, not including implicit H atoms. || ||x = {{mol1}}.find({{mol2}}, "map"+flags,format) | Creates a correlation map of atoms of mol1 to atoms of mol2. Similar to compare({{mol1}},{{mol2}},"map", options) but more flexible in terms of return value. Creates a correlation map of atoms of mol1 to atoms of mol2, for example {{1.1}} and {{2.1}}. Uses SMILES; Jmol SMILES directive flags such as "hydrogen" or "open" can be added. The format to use for the results can be "name", "index", "number" or any valid label, such as "%a %i", defaulting to "number". Returns a map (an associative array) containing keys: [|| smiles | the SMILES string used for the match || BS1 | atomset for mol1 || BS2 | atomset for mol2 || SMILEStoBS1 | array correlating SMILES atoms to BS1 || SMILEStoBS2 | array correlating SMILES atoms to BS2 || BS1toBS2 | array correlating BS1 to BS2 (indexes are for mol1; values are mol2) || MAP1to2 | array of [a,b] pairs, where a is format for mol1, b is format for mol2 || key | format used in mapping ||] For example: :> :> load files ":caffeine" "$caffeine" :> info = {{1.1}}.find({{2.1}},"map", "name") :> print info.SMILES :> print info.key + ": " + info.MAP1to2.format("JSON") :> :>> O=C1c2c3N(C)C(=O)N1C.[n]2(C)c[n]3 :> :>> name: [ [ "O1","O14" ],[ "O2","O4" ],[ "N3","N5" ],[ "N4","N10" ],[ "N5","N1" ], :>> [ "N6","N8" ],[ "C7","C12" ],[ "C8","C7" ],[ "C9","C13" ],[ "C10","C3" ], :>> [ "C11","C9" ],[ "C12","C6" ],[ "C13","C11" ],[ "C14","C2" ] ] :> :> info = {{1.1}}.find({{2.1}},"map hydrogen", "number") :> print info.SMILES :> print info.key + ": " + info.MAP1to2.format("JSON") :> :>> /hydrogen/O=C1c2c3N4[C@@]([H])([H])[H].N51[C@@]([H])([H])[H].C54=O.[n]62[C@]([H])([H])[H].c6([H])[n]3 :> :>> number: [ [ 1,14 ],[ 2,4 ],[ 3,5 ],[ 4,10 ],[ 5,1 ],[ 6,8 ],[ 7,12 ], :>> [ 8,7 ],[ 9,13 ],[ 10,3 ],[ 11,9 ],[ 12,6 ],[ 13,11 ],[ 14,2 ],[ 15,21 ], :>> [ 16,18 ],[ 17,20 ],[ 18,19 ],[ 19,22 ],[ 20,23 ],[ 21,24 ],[ 22,15 ],[ 23,17 ],[ 24,16 ] ] :>:> ||x = {{atoms}}.find(smartsString, "map")| Returns an array of arrays of atom indices (0-based) that indicates the exact correlation between atomIndex and position in the SMARTS string. Only unique sets of atoms are found, not every possibility. In this example we create a 1:1 mapping of atoms for the caffeine model from two sources -- NIH/CADD, and PubChem -- by correlating them both to the same SMILES string.
:>load $caffeine:>s = show("smiles") // N1(C)C(=O)c2c3N(C)C1=O.[n]2(C)c[n]3:>print {{*}}.find(s,"map").format("JSON"):>>[ [ 0,1,12,13,11,6,4,5,2,3,9,10,8,7 ] ]:>:>load :caffeine:>print {{*}}.find(s,"map").format("JSON"):>>[ [ 4,13,8,0,6,7,2,11,9,1,3,12,10,5 ] ]
||x = array.find()|returns an array that has been cleared of all "empty" elements -- ones that are empty strings, empty arrays, or empty associative arrays. For example, [0 3 4 "" 5 {{}} [] 6].find() returns [0 3 4 5 6].||x = array.find("")|returns an array that has been cleared of all "nonempty" elements -- ones that are not empty strings, empty arrays, or empty associative arrays. For example, [0 3 4 "" 5 {{}} [] 6].find("") returns ["" [] {{}}]. ||x = array.find(ns)|where ns is not a string value. Returns an array that points to the positions of array that are equal to ns. For example, [11,1,2,3,1].find(1) == [2,5]. ||x = array.format(headings) |Transform an array of arrays into an array of associative arrays based on "column" headings, and vice-versa. For example, if y = [[1,2],[3,4]], then z = y.format(["a","b"]) will set z to [ {{ "a": 1,"b": 2 }},{{ "a": 3, "b": 4 }} ]. And, in reverse, z.format(["a","b"]) will be back to [[1,2],[3,4]]||x = array.format("JSON")|Formats a serial or associative array as JSON.||x = array.format("%5.3f\t%5s")|Formats an array or array of arrays into a multiline string using sprintf format. See also the format(sprintfFormat, a, b, c) description. ||x = array.getProperty(key) |same as getproperty(array, key); looks in associative array elements of array to return an array of values associated with key.||x = {{atomset}}.getProperty("xxx") or x = {{atomset}}._("xxx")| shortcut for getProperty("atominfo.xxx", {{atomset}}). For example:
print {{*}}._("visible")
print {{*}}._("coord")
A list of available keys xxx can be shown using print @1._(). "[SELECT ... WHERE ...]" syntax can be used. ||x = $id.getProperty(intoType)| retrieve a property of type infoType for shape with ID specified. The specific information available depends upon the shape. ("+meshCollection" here means "and also those listed under meshCollection"): [||CGO| command +meshCollection ||draw| command type +meshCollection ||isosurface| area atoms colorEncoder command contours cutoff dataRange dataRangeStr jvxlDataXml jvxlFileInfo message minMaxInfo moLinearCombination moNumber nSets plane pmesh value volume +meshCollection ||measures| count countPlusIndices info infostring pointInfo stringValue ||meshCollection| bsVertices count data ID info list values vertices ||polyhedra| atomIndex atomName atomNames atomNumber bsFlat center collapsed color colorEdge element elemNos energy face_areas face_points face_types faceCount faces faceTriangles id info modelIndex modelNumber normals offset pointGroup pointGroupFamily pointScale polySmiles r scale smarts smiles triangleCount triangles vertexCount vertexIndices vertices volume ||] ||x = array.join()|Makes a flat array copy of a multidimensional array. creating an array of dimension 1. For example, if a = [1,2,[3,4,5],6], then a.join() will be [1,2,3,4,5,6]. ||x = x.join(y)|For string x, joins lines of x using the string equivalent of y into one line. For array x and string y , joins elements of x into one string, separating elements of x by s. For array x and array y , joins the two arrays into an array of arrays. (Adding a new column, effectively). If x is already an array of arrays or y is already an array of arrays, carries out an add operation on pairs of elements to maintain one array per element of the resulting array. If both x and y are arrays, the resultant array has the minimum number of elements common to x and y. For example, if x = [1, 2, 3] and y = [4, 5, 6], then x.join(y) = [ [1, 4], [2, 5], [3, 6] ], and x.join(y).join(y) = [ [1, 4, 4], [2, 5, 5], [3, 6, 6] ]. Note that x.join(y) is particularly valuable when tables of data are ultimately desired, but one does not want to go immediately into strings using x.add(s,y) (see above).||x = y.join("", TRUE)|joins an array of arrays into line-delimited comma separated value (CSV) data. Reversed using y.split("", TRUE). ||x = y.join("\\t", TRUE)|joins an array of arrays into line-delimited tab separated data.||x = y.label(format)|A list of labels for atoms or bonds using format strings. (see {#.label~})||x = atomset.modulation(type,t)| For a given (optional) type (displacement "D", magnetic "M", occupancy "O" is implemented), returns an array of values related to one or more atoms\' modulation. For displacement and magnetic, returns a vector cooresponding to the relevant phase of the modulation, t (irrespective of the setting of {#.set_modulationscale~set modulationScale}) or -1 if not modulated. For occupancy, returns a number from 0 to 100 or, if not modulated, "NaN." If t is not given, as in @33.modulation()[1], (or, alternatively, if t >= 1E6), then the unmodulated position/magnetic spin is returned for "D" or "M" or the currently calculated occupancy for "O".||x = data1.mul(data2)|See data1.add()||x = pt1.mul3(pt2)| returns {{pt1.x*pt2.x, pt1.y*pt2.y, pt1.z*pt2.z}}, or the equivalent of taking the dot product of a coordinate in space with one in reciprocal space. If either pt1 or pt2 is not a point, reverts to simple multiplication.||x = array.pivot()|Creates a pivot table of the values in the array in the form of an associative array with keys that are the set of unique values in the array, and with values that are counts of those values. ||x = {{associative array}}.pivot() | Creates an inverted associative array, where the values of the original associative array are now keys, with values that are arrays of the original keys having given values.||x = a.pop()| array pop -- removes the last entry of an array and returns its value. If the array is empty, returns an empty string. If the array is an associative array, a.pop() empties it of all keys and returns the empty array a.||x = a.pop(key)| removes and returns from an associative array the value associated with the specified key; (same as y = a - key, but does not create a new array).||a.push(x)| array push -- adds an entry of an array and returns the array. If a and x are associative arrays, adds all entries of x to a.||a.push(key, value)| adds the specified key/value pair to an associative array.||x = y.replace()|return a cleaned-up version of y with whitespace reduced to a single space and trimmed.||x = y.replace(s1,s2)|replaces all occurances of s1 with s2 in y. If y is a number, this function first converts the number to a string, then does the replacement.||x = y.replace(chars,s,TRUE)|replaces all occurrences of each character of chars with s.||x = y.split(s)|splits y into lines by replacing all occurances of s with a new-line character and converting the string to a set of lines.||x = y.split(TRUE)|splits y into tokens based on standard white space (space, tab, line endings).||x = y.split("", TRUE)|splits line-delimited comma separated value (CSV) data into an array of arrays. y may be either a single string or an array of strings, one for each line. Reversed using x.join("", TRUE). ||x = y.split("\\t", TRUE)|splits line-delimited tab separated data into an array of arrays. y may be either a single string or an array of strings, one for each line. Reversed using x.join("\\t", TRUE). ||x = data1.sub(data2)|See data1.add()||x = y.substring()| Jmol math does not include a .substring() function. Instead, this is handled using the more general item-selector [i][j] syntax, described below.||x = a.symop(op,atomOrPoint)| Returns the point that is the result of the transformation of atomOrPoint via a crystallographic symmetry operation. The atom set a selects the unit cell and spacegroup to be used. If only one model is present, the symop() command alone, without "a." can be used. For the current model, @@1.symop() can be used. The first parameter, op, is a symmetry operation. This can be the 1-based index of a symmetry operation in a file (use show spacegroup to get this listing) or a specific Jones-Faithful expression in quotes such as "x,1/2-y,z". ||x = a.symop(op)| This form of the a.symop() function return the 4x4 rotation-translation matrix for this symmetry operator || x = a.symop(....,type) | All symop() methods allow a final parameter that determines the form of the return. Possibilities include: [|| "array"| Returns an associative array that contains critical information relating to this operation. Keys include axisVector axispoint cartesianTranslation fractionalTranslation id label matrix plane rotationAngle timeReversal xyz xyzOriginal. These keys can be used for the type as well: x = @@1.symop(3, "axisVector")||"atom" |Returns the atom that is the target of this operation. ||"draw" |Returns the Jmol script illustrating this operation with DRAW commands. These DRAW commands describe the symmetry operation visually, in terms of rotation axes, inversion centers, planes, and translational vectors. The draw objects will all have IDs starting with "draw_", which can be substituted using .replace("draw_","xxx").||"full" |Returns the tab-separated Jones-Faithful string and descriptor for this operation. For example:
:>print symop(3,"full") :>:>> -x,-y,-z(mx,my,mz) Ci: 0 0 0
|| "label" | Returns a short description of the operation, such as c-glide plane|translation: 0 0 1/2|| "lattice" |Returns the lattice type associated with the space group involving this operation. || "list" |Specifically when two atoms or points are specified, returns a string list of all operations taking the first to the second. :>:>print symop(@3,@21,"list") :>:>>5 x+1/2,-y+1/2,-z+1/2(-mx,my,mz) 2-fold screw axis|translation: 1/2 0 0|time-reversed :>>7 -x+1/2,y+1/2,z+1/2(-mx,my,mz) n-glide plane|translation: 0 1/2 1/2|time-reversed || "point" | Returns the point that is the target of this operation.||]||x = y.tensor("type","property")|returns one of several tensor properties using tensors read by certain file readers (CASTEP, QuantumEspresso, Magres). "type" might be "charge" or "isc" or "ms", for example. Eigenvectors are sorted based on their corresponding eigenvlues, using the convention: ¦v3 - v1¦ >= ¦v3 - v2¦ > ¦v2 - v1¦. Implemented "property" values include:[|| "anisotropy"|v3 - (v1 + v2)/2|| "asymmetry"|(v2 - v1)/(v3 -v_iso)|| "asymMatrix"|matrix representation of the asymmetric tensor component|| "chi"|specific to electric field gradient (efg) tensors, the quadrupolar constant || "dc" | dipolar coupling constant; tensor type ignored|| "eigenvalues"|[v1,v2,v3]|| "eigenvectors"|[V1,V2,V3]|| "eulerzxz" |[alpha,beta,gamma] for z-x-z convention|| "eulerzyz" |[alpha,beta,gamma] for z-y-z convention|| "indices" |[modelIndex,atomIndex1,atomIndex2], with atomIndex2 = -1 if this tensor is single-atom based|| "isotropy" | (v1 + v2 + v3)/3 || "j" | J-coupling constant;specific to indirect spin coupling (isc) tensors, in hz|| "quaternion" | the quaternion representing the rotation assoicated with the symmetric matrix|| "span" | abs(v2 - v0) || "skew" | (3 (v1 - isotropy) / span)|| "symMatrix" |the symmetric matrix.|| "value" | v3, the eigenvalue furthest from the mean||"string"| a listing of selected readable data|| "type" | the tensor type, such as "charge" or "isc" or "ms" ||] Note that any other property name results in the entire set of properties to be returned in an associative array. ||x = y.trim("chars")|trims any one of the characters specified from both ends of the string y, or from every line of y if y is a set of lines.||x = {{atoms}}.within(distance,[points]) | returns the subset of {{atoms}} that are within the specified distance of any point in the array of points.||x = {{atoms}}.within(distance,{{otheratoms}})| returns the subset of {{atoms}} that are within the specified distance of any atom in {{otheratoms}}. Atoms in {{otheratoms}} are excluded.||]','TEXT','x.f(y) functions','') newCmd('','11.1.49','','*v+11.2;','Specific elements of lists and strings can be selected using a square bracket notation similar to that used for {#.atom expressions~}. Positive numbers select for the specified element. For example, x = "testing"[3] selects for the third letter of the string, "s"; Zero selects for the last element: x = "testing"[0] selects "g". Negative numbers count from the end of the string backward, so x = "testing"[-1] selects "n". Two selectors select an inclusive range of items. x = "testing"[2][4] acts similar to a "substring()" method and selects characters 2 through 4 -- "est"; x = "testing"[-1][0] selects the last two characters, "ng". Similarly, for arrays, array("this", "test", 3)[0] selects the number 3, and x = array("this", "test", 3); print x[1][2] prints
this
test
','TEXT','item-selector [i][j]','') newCmd('','','','*v+11.8;','Jmol allows for user-defined functions. Functions can appear in math expressions and can also be used as stand-alone "commands" without parentheses or commas. For example, function test(a,b,c){{...}} can be used in print test(1,2,3) or as the command TEST 1 2 3, without parentheses or commas. The only difference is that the return value from the function is disarded when the function is used as a command. Functions are not part of the state, so saving a state does not save user functions. Examples follow.
[||function rotateModel {{:> rotate {{atomno=1}} {{atomno=2}} 10
}}
rotateModel|Without any parameters listed, any commands within the function declaration are processed with a single command word.|| function rotateModelX(i,j,n) {{:> a[2] = getProperty("orientationInfo.moveTo"):> var x = 10:> rotate {{atomno=i}} {{atomno=j}} @{{n + x}}:> a[2] = getProperty("orientationInfo.moveTo")
}}
a = [];rotateModelX(10,11,30, a);print a[1];|Any number of parameters may be passed into a function. The variable names are local to that function. Note that, like JavaScript, if an array is passed, it is passed "by reference," meaning that if its value is changed within the function, then that change is a global change. Variables preceded by "var" as in this example are local to that function.|| function getDistance(i,j) {{:> var d = ({{atomno=i}}.distance({{atomno=j}})*100)%0 :> return d
}}
print "the distance is " + getDistance(3,5)|The return command works as one might expect, returning the value of its expression.|| function getXplusY {{:> return _x.x + _x.y
}}
print {{atomno=3}}.getXplusY
print {{*}}.getXplusY.min
print {{*}}.getXplusY.max
print {{*.CA}}.getXplusY.all
|You can define atom selector functions. The local variable _x will represent the selected atom within the function. Such function references may include parameters in parentheses provided qualifiers such as .min, .max, or .all are not used within the function itself.||]
Functions must be defined prior to use. Similar to the JavaScript language, a function must be declared prior to its inclusion in a script command. Unlike JavaScript, function names are not case-sensitive.
The PRIVATE keyword prior to a function definition (for example, private function test(){{...}}, introduced in Jmol 14.32 and 15.2) specifies that the function is private to the script in which it is being defined. If you wish to make all functions in a script private, add the one-line comment //@private at the top of the script. All functions defined in a Jmol state are implicitly private to that state script.
You can redefine a function as many times as you wish. In addition, you can save a function\'s definition using myFunctionDef = script("show function myFunction") where function myFunction has been defined. This can be useful if a function definition is to be replaced and later returned to its original value using script inline @myFunctionDef. In JavaScript, if a function name starts with "static_", then the function defined in one app will also be defined for all HTML5 apps, not just the one running the script. ','TEXT','user-defined functions','') newCmd('','14.3.15','','*v+14.4;','You have access to several variables in scripts and functions that are being called. [||_arguments|the array of arguments delivered to this function||_argCount| number of arguments delivered to this function||_caller| an associative array listing all the local VARs defined in the calling function; note that _caller._caller is not valid. But you can define var caller = _caller, and then that will be passed to the next level of function calls. Perhaps surprisingly, these local variables to the called function are modifiable by that function even though they are local to the function or script making the call.||]','TEXT','special variables for functions','') newCmd('','12.1.16','','*v+12.0;','You can replace standard Jmol commands with commands of your own, and you can call functions just like any Jmol command, without the need for parentheses or commas. For example:
[||function measure (a,b) {{:> prompt "measuring " + a + " " + b:> measure @a @b
}}
measure {{atomno=1}} {{atomno=2}} |The function "measure" replaces the Jmol {#.measure~} command, allowing now just two parameters and displaying a message box when executed.||]','TEXT','customized command functions','') newCmd('.[Jmol Parameters]','','','*v+11.2 -- new;math','Many parameters in Jmol can be {#.set~} and may also be checked using Jmol math. ','0|1','','') newCmd('','','','*v+11.2;','TABLE1','TEXT','General Parameters','') newCmd('','','','*v+11.2;','TABLE2','TEXT','Set-Only Parameters','') newCmd('','','','*v+11.2;','TABLE3','TEXT','Deprecated Parameters','') newCmd('','','','*v+11.2;','TABLE4','TEXT','Reserved Names','') newCmd('.[using the clipboard]','','','*v+11.0 -- new;clip','Model data that has been clipped from local sources, such as a return from {http://www.rcsb.org/pdb/files/1crn.pdb~the RCSB PDB file directory}, can be loaded into Jmol directly. From the application, simply use Edit...Paste; if using the applet, simply right-click on the applet and select Show...Console. Then paste the data into the lower (input) frame and click Load.','1|2','','') newCmd('.animation','','','*v+11.0 -- adds greatly expanded frame animation control;anim;image','Sets selected animation parameters or turns animation on or off. Note that there are four distinct animation types that can be employed using Jmol: (1) files may contain multiple structures that are "played" sequencially, (2) models may contain vibrational modes that can be animated, (3) certain Jmol script commands (namely {#.move~}, {#.moveTo~}, {#.navigate~}, {#.restore~restore ORIENTATION} and {#.zoomTo~}), can create the illusion of motion over a period of time, and (4) Jmol scripts can be run through in a predefined way, involving {#.loop~} and {#.delay~}. The "animation" command only refers to method (1). See also {#.set(misc)~set backgroundModel}.','1|2|4','','') newCmd('','','','*v+11.0;','Turns on or off animation. With animation ON, the {#.frame~} range is also reset to all frames. (An implicit frame range ALL command is executed. This functionality is for backward compatibility with Chime.) If this resetting is not desired because the frame range has been set, then frame PLAY or frame PLAYREV should be used instead of animation ON.','*10','._on_off{"ON"}','') newCmd('','','','*v+11.0;','Sets the animation direction to be from last frame to first frame.','*21','DIRECTION','-1') newCmd('','13.1.13','','*v+13.2;','Applies a filter to a running animation to display only a certain set of atoms.','*30','DISPLAY','{{atom set}}') newCmd('','','','*v+11.0;','Sets the animation direction to be first frame to last frame.','*20','DIRECTION','+1') newCmd('','','','*v+11.0;','Sets the animation frames per second (maximum 50).','*40','FPS','._anim_fps') newCmd('','','','*v+11.0;','animation frames allows defining of a set of models to be displayed in any desired order. For example, anim frames [ 1 2 3 4 5 10 9 8 7 6 ] . You can specify a range of values using negative numbers. For example, the sequence "1-5, then 10-6" can also be written [ 1 -5 10 -6]. This functionality is useful, for example, when output from an IRC (intrinsic reaction coordinates) calculation is being used. See additional frame options at the {#frame~} command. ','*50','FRAMES','[ array of model numbers ]') newCmd('','','','*v+11.0;','Sets the animation mode to restart the sequence automatically when the last frame has played.','*90','MODE','LOOP') newCmd('','','','*v+11.0;','Allows for a time delay at the start and end of the loop.','*91','','LOOP','._time_delay1','._time_delay2') newCmd('','','','*v+11.0;','Sets the animation to play once through and then stop (the default mode).','*92','','ONCE') newCmd('','','','*v+11.0;','Sets the animation to play forward and back endlessly.','*93','','PALINDROME') newCmd('','','','*v+11.0;','Allows for a time delay at the start and end of the palindrome.','*94','','PALINDROME','._time_delay1','._time_delay2') newCmd('','13.1.13','','*v+13.2;','The animation MORPH option interpolates between conformations in a trajectory or between two models from different sources, generating intermediate points to display. In either case, the option requires a previous LOAD TRAJECTORY. animation MORPH creates a linear morph consisting of the designated number of representations inserted between trajectories. For example, load test.pse;animation morph 3. For two models, one might use load trajectory "test1.pdb" "test2.pdb"; animation morph 32. Note that, by itself, animation MORPH does not play the animation. To do that, it must be followed by commands such as animation mode palindrome; animation on;. The {#.animation~animation frames} and {#.animation~animation display} commands do not apply when a trajectory morph is animated. ','*95','MORPH','(integer)') newCmd('.[atom expressions]','','','*v+11.8;atoms;math;prop','In Jmol, an atom expression is a mathematical expressions that represent a collection of atoms in one or more models. Examples include
:>select on protein or water:>display add carbohydrate:>color {{ala24}} red:>zoomto {{ligand}} 0:>contact {{e20}} surface:>isosurface select {{within(5, HEM)}} only vdw map property temperature:>print "the center is at " + {{*}}.xyz
Notice that some of these commands require braces {{ }} surrounding the atom expression and some do not. The basic rule is that you can always use braces, but you must use braces when a command can take arguments that are not atom expressions. So, for example, you can also color cartoons and isosurfaces and drawn objects; you can zoomto {{ligand}} 80 as well as 0. But what you select are atoms, and only atoms can be added to a display. Most commands DO require braces. The only ones that do not are the following ten: {#.center~}, {#.define~}, {#.delete~}, {#.display~}, {#.fix~}, {#.hide~}, {#.restrict~}, {#.select~}, {#.subset~}, and {#.zap~}. These are the only commands in Jmol that act specifically on atom sets.
All terms in and atom expression can be preceeded by the keyword NOT, joined by AND, OR, or XOR, and surrounded by parentheses. Terms in atom expressions include:
[||general terms|[||all| all atoms; same as *||bonded| covalently bonded||clickable| actually visible -- having some visible aspect such as wireframe, spacefill, or a label showing, or the alpha-carbon or phosphorus atom in a biomolecule that is rendered with only cartoon, rocket, or other biomolecule-specific shape. ||connected| bonded in any way, including hydrogen bonds||displayed| displayed using the {#.display~} or {#.hide~} command; not necessarily visible||hidden| hidden using the {#.display~} or {#.hide~} command||none| no atoms||selected| atoms that have been selected; defaults to all when a file is first loaded||solvent| PDB "UREA", "SO4", "PO4", "HOH", "DOD", "WAT"; in any model the connected set of H-O-H||thisModel| atoms in the current frame set, as defined by {#.frame~}, {#.model~}, or {#.animation~} commands. If more than one model is in this set, "thisModel" refers to all of them, regardless of atom displayed/hidden status.||visible|visible in any way, including PDB residue atoms for which a cartoon or other such rendering makes their group visible, even if they themselves are not visible.||]
||file.model|as, for example, select 3.2, a specific model in a specific file. Note that select 3.0 selects all atoms in all models of the third file of the most recent {#.load~}. ||subset|the currently defined {#.subset~}. Note that if a subset is currently defined, then select/display all is the same as select/display subset, restrict none is the same as restrict not subset. In addition, select not subset selects nothing. ||specialPosition|atoms in crystal structures that are at special positions - that is, for which there is more than one operator that leads to them.||symmetry-related|These selections relate to the unit cell and space group symmetry. See also {#.atomproperties~atom properties} fx, fy, fz, fxyz, ux, uy, uz, uxyz, and symop. [||symmetry|all atoms that are derived from the asymmetric unit (!symmetry or symop=1555) by symmetry operations or lattice translations||unitcell|atoms of the current {#.unitcell~}, which may be offset. this includes atoms on all faces, edges, and vertices of the unitcell||within(unitcell)|atoms "within" the current {#.unitcell~} -- that is, atoms with fractional coordinates ≥0 and <1||]|| polyhedra | all central atoms for which {#.polyhedra~} have been created. See also polyhera(n), below. ||chemical elements|element_name (including "deuterium and tritium"), _Xx (an element symbol preceeded by underscore, possibly with isotope number listed, such as _Cu, _Fe, _2H, _31P)||terms based on the periodic table of the elements| [||nonmetal | _H,_He,_B,_C,_N,_O,_F,_Ne,_Si,_P,_S,_Cl,_Ar,_As,_Se,_Br,_Kr,_Te,_I,_Xe,_At,_Rn||metal | !nonmetal||alkaliMetal| _Li,_Na,_K,_Rb,_Cs,_Fr||alkalineEarth | _Be,_Mg,_Ca,_Sr,_Ba,_Ra||nobleGas | _He,_Ne,_Ar,_Kr,_Xe,_Rn||metalloid | _B,_Si,_Ge,_As,_Sb,_Te||transitionMetal | (includes La and Ac) elemno>=21 and elemno<=30, elemno=57, elemno=89, elemno>=39 and elemno<=48, elemno>=72 and elemno<=80, elemno>=104 and elemno<=112||lanthanide | (does not include La) elemno>57 and elemno<=71||actinide | (does not include Ac) elemno>89 and elemno<=103||]||isaromatic|atoms connected with the AROMATIC, AROMATICSINGLE, or AROMATICDOUBLE bond types||non-protein/nucleic groups|carbohydrate, hetero, ions (specifically the PDB designations "PO4" and "SO4"), ligand (originally "hetero and not solvent"; changed to "!(protein,nucleic,water,UREA)" for Jmol 12.2), sidechain||nucleic acid residues|[||nucleic| any group that (a) has one of the following group names: G, C, A, T, U, I, DG, DC, DA, DT, DU, DI, +G, +C, +A, +T, +U, +I; or (b) can be identified as a group that is only one atom, with name "P"; or (c) has all of the following atoms (prime, \', can replace * here): C1*, C2*, C3*, O3*, C4*, C5*, and O5*.||purine| any nucleic group that (a) has one of the following group names: A, G, I, DA, DG, DI, +A, +G, or +I; or (b) also has atoms N7, C8, and N9.||pyrimidine| any nucleic group that (a) has one of the following group names: C, T, U, DC, DT, DU, +C, +T, +U; or (b) also has atom O2.||dna| any nucleic group that (a) has one of the following group names: DG, DC, DA, DT, DU, DI, T, +G, +C, +A, +T; or (b) has neither atom O2* or O2\'.||rna| any nucleic group that (a) has one of the following group names: G, C, A, U, I, +U, +I; or (b) has atom O2* or O2\'.||]||protein residues|[||protein| defined as a group that (a) has one of the following group names: ALA, ARG, ASN, ASP, CYS, GLN, GLU, GLY, HIS, ILE, LEU, LYS, MET, PHE, PRO, SER, THR, TRP, TYR, VAL, ASX, GLX, or UNK; or (b) contains PDB atom designations [C, O, CA, and N] bonded correctly; or (c) does not contain "O" but contains [C, CA, and N] bonded correctly; or (d) has only one atom, which has name CA and does not have the group name CA (indicating a calcium atom). ||acidic|ASP GLU||acyclic| amino and not cyclic||aliphatic|ALA GLY ILE LEU VAL||amino|all twenty standard amino acids, plus ASX, GLX, UNK||aromatic|HIS PHE TRP TYR (see also "isaromatic" for aromatic bonds)||basic|ARG HIS LYS||buried| ALA CYS ILE LEU MET PHE TRP VAL||charged|same as acidic or basic -- ASP GLU, ARG HIS LYS||cyclic|HIS PHE PRO TRP TYR||helix, helixalpha, helix310, helixpi|secondary structure-related. ||hetero|PDB atoms designated as HETATM||hydrophobic| ALA GLY ILE LEU MET PHE PRO TRP TYR VAL||large |ARG GLU GLN HIS ILE LEU LYS MET PHE TRP TYR||medium|ASN ASP CYS PRO THR VAL||negative|same as acidic -- ASP GLU||neutral| amino and not (acidic or basic)||polar| amino and not hydrophobic||positive|same as basic -- ARG HIS LYS||sheet|secondary structure-related||small|ALA GLY SER||surface| amino and not buried||turn|secondary structure-related||]||protein/nucleic acid-related|[||alpha |(*.CA)|| base |nucleic acid bases||backbone (or "mainchain")|(*.C, *.CA, *.N, and all nucleic other than the bases themselves)|| sidechain |((protein or nucleic) and not backbone)|| spine |(*.CA, *.N, *.C for proteins; *.P, *.O3\', *.O5\', *.C3\', *.C4\', *.C5 for nucleic acids)||leadatom |(*.CA, *.P, and terminal *.O5\')||] ||DSSR structure|Available after load =xxxx/dssr. [||bulges| within(dssr,"bulges") ||coaxStacks| within(dssr,"coaxStacks") ||hairpins| within(dssr,"hairpins") ||hbonds| within(dssr,"hbonds") ||helices| within(dssr,"helices") ||iloops| within(dssr,"iloops") ||isoCanonPairs| within(dssr,"isoCanonPairs") ||junctions| within(dssr,"junctions") ||kissingLoops| within(dssr,"kissingLoops") ||multiplets| within(dssr,"multiplets") ||nonStack| within(dssr,"nonStack") ||nts| within(dssr,"nts") ||naChains| within(dssr,"naChains") ||pairs| within(dssr,"pairs") ||ssSegments| within(dssr,"ssSegments") ||stacks| within(dssr,"stacks") ||stems| within(dssr,"stems") ||] ||]
The comparison operators <, <=, =, >, >=, !=, and LIKE operate with many keywords. Note that = and != are not case-sensitive in Jmol, so select group="ARG" is the same as select group="arg". If you want to compare cases, use LIKE: select chain like "D". See {#.atomproperties~atom properties}. ','0','','') newCmd('','11.1.14','','*v+11.2;','An atom expression is simply a list of atoms. You can select a single atom or a range of atoms from an atom expression. The way to do this is simply to suround the atom expression with parentheses and follow it with one or two numbers in brackets: select (carbon)[3][5].This says, "Select the third through fifth carbon atoms." If the second selector is not present, then only a single atom is selected; the selector [0] indicates the last atom in the set, and negative numbers count back from that atom. Thus, select (*)[0] selects the last atom, and select (carbon and 2.3)[-1][0] selects the last two carbon atoms in model 2.3. Atom selectors can be used for any expression embedded in another command. In that case an additional set of parentheses or braces is required around the whole expression: measure {{(_O)[1]}} {{(_O)[2]}}.','TEXT','Atom selectors','') newCmd('','11.1.12','','*v+11.0;','The following functions are also supported for the selection and specification of atom sets:[||CONNECTED()| allows for selection of specific atoms based on their connectivity to other atoms. The general format is: connected([optional min # bonds], [optional max # bonds], [optional bond type], [optional atom expression]) Bond type may be any described for {#.connect~}. See {misc/groups.txt~groups.txt} for many examples of using connected() with {#.define~} || POLYHEDRA(n) | all central atoms for polyhedra with n vertices. || SEARCH(pattern)| selects atoms that match {http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html~SMARTS pattern} such as "[C,N]" or "a". The pattern may be prefixed with any Jmol SMILES directives as described in {#.jmolsmarts/smiles~Jmol SMARTS/SMILES}.|| SMILES(smiles)| selects atoms if and only if there is only one model loaded and that model matches the {http://www.daylight.com/dayhtml/doc/theory/theory.smiles.html~SMILES string} such as "CCC" (propane) or "c1ccccc1" (benzene). The pattern may be prefixed with any Jmol SMILES directives as described in {#.jmolsmarts/smiles~Jmol SMARTS/SMILES}.||WITHIN(setName,atomExpression)|any atom within a given set. The setName can be any one of the words BOUNDBOX, CHAIN, ELEMENT, GROUP, MODEL, MOLECULE, POLYMER, SITE, or STRUCTURE, or it can be a protein or nucleic acid sequence expressed in single-letter notation surrounded by quotation marks as, for example, "GGCCCTT" or "MAACYXV" (in which case the sequence is found within the expression). (SITE refers to all crystallographic sites common to the specified atom set; BOUNDBOX refers to the smallest box containing the atom set.) Additional options, including "BASEPAIR", "SMILES", and "SMARTS", are discussed below.||WITHIN(distance, withinAllModels, atomExpression)|any atom within the specified distance of any atom in the atomExpression. The optional TRUE/FALSE flag withinAllModels (by default FALSE) may be set TRUE to allow finding atoms in one model that may be within some distance of another model. If the distance is negative, then the operation applies to all atoms having normalized unit cell coordinates within the given distance of the designated atoms. If withinAllModels is TRUE, matching atoms within atomExpression itself are excluded.|| WITHIN(distance, {{x y z}})|any atom within the specified distance of the given fractional or Cartesian coordinate. If the distance is negative, then the operation applies to all atoms having normalize unit cell coordinates within -distance of the designated atoms.|| WITHIN(distance, $surfaceObject) | any atom within the given distance of a point on the specified surface. || WITHIN(x.x, VDW, {{atomset}})| Selects atoms that have overlapping van der Waals surfaces with {{atomset}}. The distance parameter is optional, and its meaning depends upon its magnitude. If it is > 10, then it is assumed to be a percent, such as 110%, 100%, 90%; otherwise it is assumed to be a distance to be added to the van der Waals radii of both atoms under consideration. So, for example, select GROUPS within(1.4, VDW, {{*:A}}) selects all groups with solvent-accessible surfaces overlapping with chain A, and select GROUPS within(100, VDW, {{ligand}}) selects all groups "clashing" with any ligand.||WITHIN(nResidues,GROUP,{{atoms}})| groups that are within a given number of residues of a specified group of atoms. || WITHIN(0,planeType, planeDesignation)| selects for any atoms within 0.01 Angstroms of a plane. If planeType is HKL, then planeDesignation is in the form {{h k l}}, where h, k, and l are Miller indices. If planeType is PLANE, then planeDesignation should be of the form @{{plane(a,b,c)}}, where a, b, and c are atom expressions or coordinates. ||WITHIN(distance,planeType, planeDesignation)|selects for atoms within the given distance in Angstroms from the plane. Positive distances are on one side; negative distances are on the other side. Experimentation may be necessary to determine which side is which for these purposes. In all cases the atoms in the plane itself (within 0.01 Angstroms of the plane on either side) are included. ||WITHIN(distance,$P)|where $P is a {#.polyhedra~polyhedron} ID. Options include on or within the polyhedron (distance == 0), angstroms within the polyhedron (distance < 0), and within distance angstroms of the polyhedron (distance > 0).||WITHIN(ATOMNAME,"aa,bb,ccc")|any atom having a listed atom name ||WITHIN(ATOMTYPE, "atomType,atomType,..."| selects for atoms of one or more atom type. Atom type is defined in certain file types, including MOL2 model files and AMBER topology files. For other file types, atom types are the same as atom names. For example, select within(ATOMTYPE,"HW,OW") selects all water atoms an AMBER topology file.||WITHIN(BASEPAIR("XY...")| finds all atoms within hydrogen-bonded DNA or RNA basepairs. Any number of pairs can be indicated. For example, display within(BASEPAIR,"GCAU") would select only G-C and A-U pairs. (Note that the RasMol-derived predefined sets "gc" and "at" refer simply to "G or C" and "A or T", respectively, and do not relate to base pairing.) || WITHIN(BOUNDBOX)| selects all atoms within the currently defined {#.boundbox~}||WITHIN(BRANCH,{{first atom}}, {{second atom}})| selects the second atom and all atoms in the molecular branch starting with the second atom but not including the first atom.||WITHIN(DSSR,type)| For more information on this function, see {misc/JmolSQLforDSSR.pdf~JmolSQLforDSSR.pdf}. Provided DSSR information is loaded, this function selects atoms within the given type of secondary structure of RNA or DNA. Options include bulges, coaxStacks (coaxial stacks), hairpins, hbonds, helices, iLoops (internal loops), isoCanonPairs (isocanonical pairs), junctions, kissingLoops, isolatedPairs, multiplets, nonStack, nts (nucleotides), pairs, ssSegments (single-stranded segments), stacks, and stems. For example, the following script produces a "3D stem diagram" for a nucleic acid:
:>load =1msy/dssr:>set cartoonSteps:>backbone -0.2:>select within(dssr,"helices"):>color blue :>select within(dssr,"stems"):>color red :>select within (dssr,"ssSegments"):>color white:>select within (dssr,"multiplets"):>color green:>select within (dssr,"isolatedPairs"):>color orange:>select leadatom:>spacefill 1.5:>label $P%[group1]:>font label 24 bold:>set labeloffset 0 0:>color label grey
In addition, the type may be followed by one of the following:[||.n| where n is a number starting with 1, to designate a specific region of that type: select within(dssr,"bulges.3")||[SQL phrase] | to be more specific: select within(dssr,"pairs[WHERE name != \'WC\']")||]
||WITHIN(HELIX)|Selects groups that would be selected using select helix but are not at either end of a helix section.||WITHIN(SEQUENCE,"sequence")| a protein or nucleic acid sequence expressed in single-letter notation surrounded by quotation marks as, for example, "GGCCCTT" or "MAACYXV" (the entire sequence must be found; as indicated above, the keyword SEQENCE is optional). || WITHIN(SHEET)|Selects groups that would be selected using select sheet but are not at either end of a sheet section. || WITHIN(SMARTS,"smartsString") | all atoms that conform to the given SMARTS string are found. When used as a math function, this method returns a list of all matching sets of atoms; when used in a selection context (SELECT, DISPLAY, HIDE, etc.), all matching atoms are returned. Only hydrogen atoms that are explicitly indicated as [H] are returned. Extensive details on Jmol 3D-SEARCH SMARTS capability may be found {http://jmol.svn.sourceforge.net/viewvc/jmol/trunk/Jmol/src/org/jmol/smiles/package.html~on-line}. || WITHIN(SMILES,"smilesString") | all atoms that conform to the given {http://www.daylight.com/dayhtml/doc/theory/theory.smiles.html~SMILES} string are found. When used as a math function, this method returns a list of all matching sets of atoms, including any indicated hydrogen atoms or hydrogen atoms required to complete the valence on an atom. When used in a selection context (SELECT, DISPLAY, HIDE, etc.), all matching atoms are returned. Note that for substructure searches, WITHIN(SMARTS,"smartsString") is recommended. ||WITHIN(unitid) | all atoms matching the given {https://www.bgsu.edu/research/rna/help/rna-3d-hub-help/unit-ids.html~Unit ID}. Unit IDs consist of nine fields separated by |:
:>pdbid|model|chain|RESNAME|resno|ATOMNAME|altcode|inscode|symmetry:>
They can be truncated on the right, but not the left: 1ehz|1|A|G|15, |1|A|G|15|C2. At a minimum, they require model number, chain, and residue number: |1|A||15. A blank ATOMNAME field specifies a full residue, with altcode indicating "this alt_id or no alt_id" (a configuration). A nonblank ATOMNAME field specifies one atom, with altcode indicating "exactly this alt_id" (a location). Unit IDs will be identified by their signature, disregarding white space, commas, "]", "[", and double quote: select within("1ehz|1|A|G|15||||,1ehz|1|A|U|59||||,1ehz|1|A|C|60|||");||]','TEXT','Functions','') newCmd('','','','*v+11.0;','The general specification of atoms in PDB "residues" follows the method used in RasMol. While the order of specifiers is somewhat flexible, the following order is generally applicable:
[residueType]seqRange ^insertionCode :chainLetter .atomName %altLoc /modelNumber
[||[residueType]|[ALA] [G] [2E1] [L??] When used without any other specifiers it is possible in some but not all cases to leave off the brackets around the residue type. However, leaving off the brackets is not recommended and is known to fail when the residue type begins with a number.||seqRange| 1 1-30 40- Note that ranges refer to physical ranges of data in the file. If residues corresponding to both the starting and ending residue numbers are not present in the file, selection returns no atoms. If residues with numbers between the starting and ending numbers are out of place in the file -- not physically between those two file positions -- they will not be included in the selection. If there is a desire to include such residues, or the selection should allow starting or ending residues to not be present, then use the resno comparison method instead. In this case, for example: select resno >=1 and resno <=30 or select resno >= 40. ||^insertionCode | ^A ^B ^? ||:chainLetter | :A :B :? ||.atomName |.Ca .C? .? .?? ||%altLoc | %1 %A %? ||/modelNumber| /1 /2 /* refer to the number on the MODEL record in multimodel PDB files or the sequential number of the model in the file otherwise. When multiple files are loaded, these numbers refer to the file number, indicating "all models in that file." Specific models in specific files can be specified using a decimal notation: file.model as, for example, select *.CA/2.1 -- all alpha carbons in the first model of the second file listed in the {#.load~} command.||]','TEXT','RasMol biomolecular residue specifications','') newCmd('','','','*v+11.0;','Unspecified components of the atom specification are indicated in some cases using a question mark and in others using an asterisk.
For PDB files specifically, the wildcard * can be used in place of [residueType]seqRange to indicate "any." For example: select *.CA. Wildcards can be used elsewhere in the specification, but it is preferred simply to not include a specifier altogether. Thus, select [ALA].* is the same as select [ALA]. Note that in the case of PDB files and MOL2 files with residues indicated, * may be used in the form x* only in the case of residue names, not atom names. Thus, select AS* selects aspartate and asparagine. When used for an atom, for example, with the unremediated PDB file 1bkx select A.O?* the * is not wild and selects atoms A.O1* and A.O4*. (In remediated PDB files, this * becomes a single quote or "prime" character -- AO1\', AO4\'.) Asterisks cannot be used in place of insertionCode or altLoc.
Question marks are used to indicate "some character": select *.C??. Note that the number of question marks is significant. ".?" only finds atoms with single-letter names such as "O" and "C"; ".??" finds atoms with single-letter or double-letter names. The specification :?, ^?, and %? mean SOME chain, SOME insertion code or SOME alternate location; use :, ^, and % alone to indicate "atoms without chain indication," "atoms without insertion code," and "atoms without alternate location," respectively.
You can use \\? to match an actual ? in an atom name. For instance, if there are two atoms, one with the name "O1" and one with the name "O1?" then select O1? will select both atoms, but select O1\\? will select only the second atom. You cannot use \\* to escape an actual * in an atom name.
For other file types, ? can be used anywhere in an atom name, and * can be used at the end of a name. To use * at the beginning of a name, use ?*, not just *.
General matching of atom names is not case sensitive. Note that the LIKE operator allows full support for standard use of ? and *, and it is case sensitive. So, for example: search atomName LIKE "*15".','TEXT','Wildcards','') newCmd('','','','*v+11.0;','Atom names can also be used for some non-PDB file types. For example, in CIF files, the atom_site_label field is used for the atom name. If an atom has the label "C34" you can select it using select *.C34 or select C34 or even select C*. Note that in this case, the wildcard * is no problem, since non-PDB file types do not include residue names, which might conflict with atom names. Similarly, Jaguar, NWChem, Tripos MOL2, Wavefunction Odyssey, SHELX, and Wavefunction Spartan files list atoms as "H3" and "O2". Atoms for these file types can be selected using these names, and the names can be displayed in {#.label~labels} using the format code %a. For file types such as XYZ that do not indicate a number with the atom symbol, Jmol constructs an atom name from the element symbol and the sequential number of the atom in the file.','TEXT','Atom names for other file types','') newCmd('.[atom properties]','','','*v+11.8;prop;math','Over 100 atom properties can be selected or retrieved from model data, and most of these can be set as well. The older, more limited Rasmol notation [group].atomName^insertion:chain%altloc can still be used, but equally well one can combine any subsets of those using a more natural notation. For example, select group="ARG" and atomname="CA" and chain="A" is equivalent to select [ARG].CA:A. Mostly, the newer xxxx=y notation generalizes better in terms of additional parameters not unique to Rasmol. The comparison operators <, <=, =, >, >=, !=, and LIKE operate with many keywords. Note that = and != are not case-sensitive in Jmol, so select group="ARG" is the same as select group="arg". If you want to compare cases, use LIKE: select chain like "D". If you want to do case-sensitive string comparisons, use LIKE: select chain like "D". Note that LIKE is only for string comparison. select atomono like 3 returns nothing.
Atom properties may also be set directly using {{atom expression}}.xxxx = y. For example, {{_H}}.radius = 0 or {{*}}.radius = for(x;{{*}};x.adpmax - x.adpmin) or {{*}}.radius = {{*}}.temperature.all.mul(0.03). An associative array of all matching atom properties can be created using {{atom expression}}.xx?, where "xx" is the beginning of a property name, or even just {{atom expression}}.? for all atom properties. (for example, print {{atomno=3}}.a? or print @3.?).
Starting in Jmol 14.2, you can apply operations to arrays of values. For example, select atomno=[1,2,4] or display color !=["red","white"]. These can be read "select atoms for which the atomno is 1, 2, or 4" and "display all atoms for which the color is not red or white."
For {#.label~labels}, the %[xxx] notation extends the original single-character %x code of early Jmol versions. For example, label %[atomno] or label %8.2[xyz]. Within labels, %% will be interpreted as "%" to avoid its being treated as a special character. This is only needed in the rare case where its context could be interpreted as a variable token, for example, in label 30%x, if that needed to be displayed exactly like that for some reason.
The full list of atom properties is given below.
[||property|select xxx=y|label %[xxx]|label %x|print {{*}}.xxx|
[|| 0 | noGroup|| 1-5 |ALA, ARG, ASN, ASP, CYS|| 6-10| GLN, GLU, GLY, HIS, ILE|| 11-15| LEU, LYS, MET, PHE, PRO|| 16-20 | SER, THR, TRP, TYR, VAL || 21-23 | ASX, GLX, UNK|| 24-29| A, +A, G, +G, I, +I || 30-35| C, +C, T, +T, U, +U||]
Additional unique numbers are assigned arbitrarily by Jmol and cannot be used reproducibly. ||property|select xxx=y|label %[xxx]|label %x|print {{*}}.xxx|{{*}}.xxx = y|description||groupindex|yes|yes|%G|yes| | overall group index ||hydrophobicity| yes | yes | | yes | | Aminoacid residue scale of hydrophobicity based on {http://www.ncbi.nlm.nih.gov/pubmed/4023714~Rose, G. D., Geselowitz, A. R., Lesser, G. J., Lee, R. H., and Zehfus, M. H. (1985). Hydrophobicity of amino acid residues in globular proteins, Science, 229(4716):834-838}.||identify|yes|yes|%U|yes| |for a PDB/mmCIF file, a label such as [ILE]7^1:A.CD1%A/3 #47, which includes the group ([ILE]), residue number with optional insertion code (7^1), chain (:A), atom name (CD1), alternate location if present (%A), PDB model number (/3, for NMR models when one file is loaded; /file.model such as /2.3 if more than one file is loaded), and atom number (#47). For non-PDB data, the information is shorter -- for example, H15/2.1 #6, indicating atom name (H15), full file.model number (/2.1), and atom number (#6). If only a single model is loaded, %[identify] does not include the model number.||insertion|yes|yes|%E|yes| |protein residue insertion code||label|yes|yes| |yes|yes|current atom label (same as format)||mass|yes|yes| |yes| |atomic mass -- especially useful with appended .max or .sum||model|yes|yes|%M|yes| |model number||modelindex|yes|yes| |yes| |a unique number for each model, starting with 0 and spanning all models in all files||||modO| |yes| |yes| |currently calculated occupancy from modulation (0 to 100; NaN if atom has no occupancy modulation)||modXYZ| |yes| |yes| |currently calculated displacement modulation (for incommensurately modulated structures). Also modX, modY, modZ for individual components. For atoms without modultion, {{xx}}.modXYZ is -1 and {{xx}}.modX is NaN, and in a label %[modXYZ] and %[modX] are blank.||molecule|yes|yes|%N|yes| |molecule number||monomer|yes|yes|%g|yes| |monomer number (group number) in a polymer (usually a chain), starting with 1, or 0 if not part of a biopolymer -- that is, not a connected carbohydrate, amino acid, or nucleic acid (Jmol 14.3.15)||ms|yes|yes| |yes| |magnetic shielding calculated from file-loaded tensors.||nbo| |yes| | | |a resonance structure configuration found in an NBO .46 or .nbo file; requires the previous issuing of {#.connect~connect NBO}.||occupancy|yes|yes|%q/%Q|yes|yes| CIF file site occupancy. In SELECT command comparisons ("select occupancy < 90"), an integer n implies measurement on a 0-100 scale; also, in the context %[occupancy] or %q for a label, the reported number is a percentage. In all other cases, such as when %Q is used in a label or when a decimal number is used in a comparison, the scale is 0.0 - 1.0.||partialCharge|yes|yes|%P|yes|yes|partial charge||phi|yes|yes|%f|yes| |protein group PHI angle for atom\'s residue ||polymer|yes|yes| |yes|yes|sequential polymer number in a model, starting with 1.||polymerLength|yes|yes|%L|yes| |polymer length||property_xx|yes|yes| |yes|yes|a property created using the DATA command||psi|yes|yes|%p|yes| |protein group PSI angle for the atom\'s residue ||radius|yes|yes| |yes|yes|currently displayed radius -- In SELECT command comparisons ("select radius=n"), integer n implies Rasmol units 1/250 Angstroms; in all other cases or when a decimal number is used, the units are Angstroms.||property|select xxx=y|label %[xxx]|label %x|print {{*}}.xxx|{{*}}.xxx = y|description||resno|yes|yes|%R|yes| |PDB residue number, not including insertion code (see also seqcode, below)||selected|yes| | |yes|yes|1.0 if atom is selected; 0.0 if not ||sequence|yes|yes| |yes| |PDB one-character sequence code, as a string of characters, with "?" indicated where single-character codes are not available||seqcode|yes|yes|%r|yes| |PDB residue number, including insertion code (for example, 234^2; "seqcode" option added in Jmol 14.3.16)||seqid|yes|yes| |yes| | (mmCIF only) the value from _atom_site.label_seq_id; a pointer to _entity_poly_seq.num in the ENTITY_POLY_SEQ category specifying the sequence of monomers in a polymer. Allowance is made for the possibility of microheterogeneity in a sample by allowing a given sequence number to be correlated with more than one monomer id. (Jmol 14.2.3)||shape|yes|yes| |yes| |hybridization geometry such as "tetrahedral"||site|yes|yes|%S|yes| |crystallographic site number||spacefill|yes|yes| |yes|yes|currently displayed radius||straightness|yes|yes|%T|yes| |quaternion-derived straightness (second derivative of the quaternion describing the orientation of the residue. This quantity will have different values depending upon the setting of quaternionFrame as "A" (alpha-carbon/phosphorus atom only), "C" (alpha-carbon/pyrimidine or purine base based), "P" (carbonyl-carbon peptide plane/phosphorus tetrahedron based), or "N" (amide-nitrogen based). The default is alpha-carbon based, which corresponds closely to the following combination of Ramachandran angles involving three consecutive residues i-1, i, and i+1: -psii-1 - phii + psii + phii+1.||strucno|yes|yes| |yes| |a unique number for each helix, sheet, or turn in a model, starting with 1.||structure|yes|yes| |yes| |The value of this parameter depends upon the context. Used with select structure=x, x can be either the quoted keyword "none", "turn", "sheet", "helix", "dna", "rna", or "carbohydrate" or a respective number 0-6. In the context {{*}}.structure, the return value is a number; in the context label %[structure], the return is one of the six keywords. ||substructure|yes|yes| |yes| |like structure, the value of this parameter depends upon the context. Used with select substructure=x, x can be either the quoted keyword "none", "turn", "sheet", "helix", "dna", "rna", "carbohydrate", "helix310", "helixalpha", or "helixpi", or the respective number 0-9. In the context {{*}}.substructure, the return value is a number; in the context label %[substructure], the return is one of the nine keywords. ||surfacedistance|yes|yes|%u|yes| |A value related to the distance of an atom to a nominal molecular surface. 0 indicates at the surface. Positive numbers are minimum distances in Angstroms from the given atom to the surface. The "surface" is not a surface, per se. It is a set of geodesic points using SET dotDensity 1;DOTS 3.0. Atoms that contribute one or more dots to this geodesic are given a value of 0; other atoms are checked for distance to each dot, and assigned that distance - 3.0.||property|select xxx=y|label %[xxx]|label %x|print {{*}}.xxx|{{*}}.xxx = y|description||sXYZ| |yes| |yes| |screen XYZ coordinates, in pixels from the left, bottom, and camera, respectively||sX|yes|yes| |yes| |screen X coordinate, in pixels from the left||sY|yes|yes| |yes| |screen Y coordinate, in pixels from the bottom||sZ|yes|yes| |yes| |screen Z coordinate, 0 at the camera, increasing with depth||symop|yes| | |yes| |the first symmetry operation code that generated this atom by Jmol; an integer starting with 1. See also symmetry, below. This operator is only present if the file contains space group information and the file was loaded using the {{i, j, k}} option so as to generate symmetry-based atoms. To select only the original atoms prior to application of symmetry, you can either use "SYMOP=n", where n is the symmetry operator corresponding to "x,y,z", or you can specify instead simply "NOT symmetry" the way you might specify "NOT hydrogen". Note that atoms in special positions will have multiple operator matches. These atoms can be selected using the keyword SPECIALPOSITION. The special form select SYMOP=nijk selects a specific translation of atoms from the given crystallographic symmetry operation. Comparators <, <=, >, >=, and != can be used and only apply to the ijk part of the designation. The ijk are relative, not absolute. Thus, symop=2555 selects for atoms that have been transformed by symop=2 but not subjected to any further translation. select symop=1555 is identical to select not symmetry. All other ijk are relative to these selections for 555. If the model was loaded using load "filename.cif" {{444 666 1}}, where the 1 indicates that all symmetry-generated atoms are to be packed within cell 555 and then translated to fill the other 26 specified cells, then select symop=3555 is nearly the same as select symop=3 and cell=555. (The difference being that cell=555 selects for all atoms that are on any edge of the cell, while symop=3555 does not.) However, the situation is different if instead the model was loaded using load "filename.cif" {{444 666 0}}, where the 0 indicates that symmetry-generated atoms are to be placed exactly where their symmetry operator would put them (x,-y,z being different then from x, 1-y, z). In that case, select symop=3555 is for all atoms that have been generated using symmetry operation 3 but have not had any additional translations applied to the x,y,z expression found in the CIF file. If, for example, symmetry operation 3 is -x,-y,-z, then load "filename.cif" {{444 666 0}} will place an atom originally at {{1/2, 1/2, 1/2}} at positions {{-1/2, -1/2, -1/2}} (symop=3555) and {{-3/2, -3/2, -3/2}} (symop=3444) and 24 other sites.||property|select xxx=y|label %[xxx]|label %x|print {{*}}.xxx|{{*}}.xxx = y|description || symmetry| |yes|%o/%O|yes| | as "symmetry" or in a label as lower-case "o" gives list of crystallographic symmetry operators generating this atom with lattice designations,such as 3555; upper-case "%O" in a label gives a list without the lattice designations. See also symop, above. ||temperature|yes|yes|%b/%t|yes|yes|temperature factor (B-factor) ||unitid| |yes| | | |{https://www.bgsu.edu/research/rna/help/rna-3d-hub-help/unit-ids.html~Unit ID}. Unit IDs consist of nine fields separated by |. unitid is allowed specfically in the %[unitid-MODE] context, where -MODE is a flag to indicate what aspects of the Unit ID to express:[|| unitid | same as unitid-ra | |1|A|G|1|OP1||| || unitid-ra | include chain, residue, and atom | |1|A|G|1|OP1||| || unitid-r | chain,residue only | |1|A|G|1|||| || unitid-mr | PDB model,chain,residue | 1EHZ|1|A|G|1|||| || unitid-mra | full unitID | 1EHZ|1|A|G|1|OP1||| || unitid-mrat | full unitID, right-trimmed | 1EHZ|1|A|G|1|OP1 ||] Note that the use of unitid is not just for display. You can also generate them using {{xxx}}.label("%[unitid]") and use them in searching {misc/JmolSQLforDSSR.pdf~DSSR data structures}. ||uXYZ| |yes| |yes| |unit cell XYZ coordinates||uX|yes|yes| |yes| |unit cell X coordinate normalized to [0,1)||uY|yes|yes| |yes| |unit cell Y coordinate normalized to [0,1)||uZ|yes|yes| |yes| |unit cell Z coordinate normalized to [0,1)||valence|yes|yes| |yes|yes|the valence of an atom (sum of bonds, where double bond counts as 2 and triple bond counts as 3||vanderwaals|yes|yes|%V|yes|yes|van der Waals radius ||vectorScale|yes|yes| |yes| |vibration vector scale|| volume|yes|yes| |yes| |approximate van der Waals volume for this atom. Note, {{*}}.volume gives an average; use {{*}}.volume.sum to get total volume. ||vXYZ| |yes|%v|yes|yes|vibration vector, or individual components as %vx %vy %vz. For atoms without vibration vectors, {{xx}}.vXYZ is -1; in a label, %[vXYZ] is blank. ||vX|yes|yes| |yes|yes|vibration vector X coordinate; for atoms without vibration vector, {{xx}}.vX is NaN (same for vY and vZ)||vY|yes|yes| |yes|yes|vibration vector Y coordinate||vZ|yes|yes| |yes|yes|vibration vector Z coordinate ||x|yes|yes|%x|yes|yes|Cartesian X coordinate ||y|yes|yes|%y|yes|yes|Cartesian Y coordinate ||z|yes|yes|%z|yes|yes|Cartesian Z coordinate ||xyz| |yes| |yes|yes|Cartesian XYZ coordinates; select xyz > 1.0 selects atoms more than one Angstrom from the origin.|| | | |%W| | |PDB residue designator with x, y, z included: [%n]%r %x %y %z ||] ','0|1','','') newCmd('.axes','','','*v+11.0 -- new;set','Turns on or off displayed axes, and determines their line style and line width (as a decimal number, in Angstroms).','0|1','','') newCmd('','','','*v+11.0;','Turns axes on or off','*1','._on_off{"ON"}','') newCmd('','','','*v+11.0;','Sets the axes diameter in Angstroms. ','*20','(decimal)','') newCmd('','12.0.RC23','','*v+12.0;','Sets the axes origin to the specified point, which may be an atom expression such as {{*}} or {{rna}}.','*21','CENTER','{{x y z}}') newCmd('','','','*v+11.0;','Sets the axes style to a thin dotted line.','*3','DOTTED','') newCmd('','','','*v+12.0;','Sets the labels for the positive X, Y, Z axes, and the origin. Negative-directed axes are hidden; empty quotes can be used to hide labels as desired.','*51','LABELS','"x-label"','"y-label"','"z-label" "origin-label"') newCmd('','13.1.12','','*v+13.2;','Sets the labels for the positive and negative X, Y, and Z axes. The origin is not labeled. ','*52','LABELS','"x-label"','"y-label"','"z-label" "-x-label" "-y-label" "-z-label"') newCmd('','','','*v+11.0;','Sets the axes diameter in pixels (1 to 19).','*4','(integer)','') newCmd('','','','*v+11.2;','Sets the axes to be based on the molecular coordinate {{0 0 0}}','*53','MOLECULAR','') newCmd('','','','*v+14.6;','For axes unitcell offsets the axes by the specified fractional coordinants in a, b, and c. Same as set axesOffset x.x and axes center {{x.x x.x x.x/1}}.','*54','OFFSET','(decimal)') newCmd('','','','','Sets the length of the axes. For historical reasons, a scale of 2.0 is "full scale." ','*620','SCALE','(decimal)') newCmd('','','','*v+14.4;','For axes position [x y] to only show specified axes.','*632','TYPE','"a" "b" "c" "ab" "ac" "bc" "abc"') newCmd('','11.9.12','','*v+12.0;ticks','Sets the parameters for ticks along the axes. An optional specific axis (X, Y, or Z) can be indicated. There are three levels of ticks - major, minor, and "subminor." Only the major ticks have labels. Which of these tick levels are displayed and the distance between ticks depends upon the parameter that takes the form of a point. This point may be in fractional form, {{1/2 0 0}}. The optional keyword FORMAT allows formating of the labels for the major ticks. These are based on an array of strings given after the FORMAT keyword. If the array is shorter than the number of ticks, the formats in the array are repeated. Following that, the optional keyword SCALE allows setting the scale either for each axis direction independently {{scaleX, scaleY, scaleZ}} or overall (as a decimal number). ','*631','TICKS X|Y|Z','{{major,minor,subminor}} FORMAT ["%0.2f", ...] ','SCALE {{scaleX, scaleY, scaleZ}} | x.x','') newCmd('','','','*v+11.2;','Sets the axes to be based on the center of the bounding box (default if not fractional coordinates)','*8','WINDOW','') newCmd('','','','*v+11.2;','Sets the axes to align with the a, b, and c axes of the unit cell (default if fractional coordinates)','*7','UNITCELL','') newCmd('','','','*v+11.8;','Sets the axes to be positioned as a small right-handed set that rotates at a specific screen coordinate or fractional position along the horizontal and vertical screen dimensions. For example, axes position [100 100]. Must be preceded by axes on. Return to standard axes using axes position OFF.','*61','POSITION','[x y] or [x y %] or OFF') newCmd('.backbone','','~structure','disp','Shows the backbone of a protein or nucleic acid macromolecule by connecting the alpha carbons. The selection of backbone units to display depends upon the currently selected atoms and the {#.set_bondmode~bondmode} setting.','0|1','','') newCmd('','','','','Turns the backbone on or off','*10','._on_off{"ON"}','') newCmd('','','','*v+11.6;','Turns backbone rendering on and all other rendering (including labels) off.','*11','ONLY','') newCmd('','','','','Backbone radius can be specified in angstroms using a decimal number (1.0, 2.0, etc.). A negative number also implies ONLY.','*2','._backbone_radius','') newCmd('.background','','','*v+11.6 -- image support;disp;color','Sets color of the background or sets the background image. For color specifications, see {#.color~}.','1|2','','') newCmd('','','','','Sets the background color of the applet/application window.','*1','._colorRGB','') newCmd('','','','','Sets the background of the most recently defined {#.echo~} text and subsequent user-defined echo text to be the given color.','*21','ECHO','._color_or_none') newCmd('','','','','Sets the background of the applet/application window to the specified image file, which can be of format BMP, JPG, PNG, or GIF. The image is stretched to fit the size of the window.','*23','IMAGE','"filename"') newCmd('','','','','Sets the background color of the atom labels that appear with the "label" command. "NONE" results in there being no label background. Operates globally, not on selected atoms.','*4','LABELS','._color_or_none') newCmd('','','','','Sets the background color for the pop-up label box that appear when the mouse "hovers" over an atom. "NONE" results in there being no hover backgrounds. Operates globally, not on selected atoms.','*3','HOVER','._color_or_none') newCmd('.bind','11.9.9','','*v+12.0;bind','The bind and {#.unbind~} commands allow users to customize the effects of mouse actions, such as a LEFT-CLICK or ALT-RIGHT-DRAG. When a mouse action is bound to a user-defined script, Jmol will replace any occurence of _ACTION, _ATOM, _BOND, _DELTAX, _DELTAY, _MODE, _POINT, _TIME, _X, and _Y in the script with appropriate values. Capitalization is significant. For example, bind "left" "doPick(_ATOM)" will execute the user\'s doPick function, supplying the expression for the atom picked.The _ACTION variable encodes the mouse/key combination involved as well as the click count (1 SHIFT, 2 CTRL, 4 RIGHT, 8 ALT or MIDDLE, 16 LEFT, 32 WHEEL all ORed with 64*clickCount). The _MODE variable indicates the mouse action that occurred (1 dragged, 2 clicked, 3 wheeled, 4 pressed, or 5 drag-released). The standard Jmol action is replaced ("unbound") by the specified user action. Definitions of (mouse action) and (Jmol action) are given below. Notice that one mouse action can have multiple bindings. The Jmol action taken depends upon the context.
[||mouse action|Jmol action|description ||LEFT+click |_assignNew |assign/new atom or bond (requires set picking assignAtom_??/assignBond_? where ?? is an element and ? is a bond order such as "SINGLE" or "DOUBLE") ||CTRL+SHIFT+LEFT+click|_center |center ||LEFT+click |_clickFrank |pop up recent context menu (click on Jmol frank) ||LEFT+click |_pickConnect |connect atoms (requires set picking CONNECT) ||LEFT+click |_deleteAtom |delete atom (requires set picking DELETE ATOM) ||LEFT+click |_deleteBond |delete bond (requires set picking DELETE BOND) ||CTRL+SHIFT+LEFT+double+drag|_depth |adjust depth (back plane; requires SLAB ON) ||LEFT+drag |_dragAtom |move atom (requires set picking DRAGATOM) ||SHIFT+LEFT+drag |_dragDrawObject |move whole DRAW object (requires set picking DRAW) ||ALT+LEFT+drag |_dragDrawPoint |move specific DRAW point (requires set picking DRAW) ||SHIFT+LEFT+drag |_dragLabel |move label (requires set picking LABEL) ||LEFT+drag |_dragMinimize |move atom and minimize molecule (requires set picking DRAGMINIMIZE) ||LEFT+drag |_dragMinimizeMolecule |move and minimize molecule (requires set picking DRAGMINIMIZEMOLECULE) ||ALT+SHIFT+LEFT+drag |_dragSelected |move selected atoms (requires set DRAGSELECTED) ||SHIFT+LEFT+drag |_dragZ |drag atoms in Z direction (requires set DRAGSELECTED) ||LEFT+drag |_navTranslate |translate navigation point (requires set NAVIGATIONMODE and set picking NAVIGATE) ||LEFT+click |_pickAtom |pick an atom ||LEFT+click |_pickIsosurface |pick an ISOSURFACE point (requires set DRAWPICKING ||LEFT+click |_pickLabel |pick a label to toggle it hidden/displayed (requires set picking LABEL) ||LEFT+click |_pickMeasure |pick an atom to include it in a measurement (after starting a measurement or after set picking DISTANCE/ANGLE/TORSION) ||CTRL+SHIFT+LEFT+click|_pickNavigate |pick a point or atom to navigate to (requires set NAVIGATIONMODE) ||LEFT+click |_pickPoint |pick a DRAW point (for measurements) (requires set DRAWPICKING ||CTRL+LEFT+down, RIGHT+down|_popupMenu |pop up the full context menu ||SHIFT+LEFT+double+click, MIDDLE+double+click|_reset |reset (when clicked off the model) ||LEFT+drag, LEFT+double+drag|_rotate |rotate ||SHIFT+LEFT+drag |_rotateBranch |rotate branch around bond (requires set picking ROTATEBOND) ||ALT+LEFT+drag |_rotateSelected |rotate selected atoms (requires set DRAGSELECTED) ||ALT+LEFT+drag, SHIFT+RIGHT+drag|_rotateZ |rotate Z ||SHIFT+LEFT+drag, MIDDLE+drag|_rotateZorZoom |rotate Z (horizontal motion of mouse) or zoom (vertical motion of mouse) ||LEFT+double+click |_select |select an atom (requires set pickingStyle EXTENDEDSELECT) ||LEFT+click |_selectToggleOr |if all are selected, unselect all, otherwise add this group of atoms to the set of selected atoms (requires set pickingStyle DRAG) ||LEFT+double+click |_setMeasure |pick an atom to initiate or conclude a measurement ||CTRL+SHIFT+LEFT+drag|_slab |adjust slab (front plane; requires SLAB ON) ||CTRL+ALT+SHIFT+LEFT+drag|_slabAndDepth |move slab/depth window (both planes; requires SLAB ON) ||LEFT+drag |_slideZoom |zoom (along right edge of window) ||LEFT+drag |_spinDrawObjectCCW |click on two points to spin around axis counterclockwise (requires set picking SPIN) ||SHIFT+LEFT+drag |_spinDrawObjectCW |click on two points to spin around axis clockwise (requires set picking SPIN) ||LEFT+double+click |_stopMotion |stop motion (requires set waitForMoveTo FALSE) ||LEFT+drag |_swipe |spin model (swipe and release button and stop motion simultaneously) ||CTRL+ALT+LEFT+drag, CTRL+RIGHT+drag, SHIFT+LEFT+double+drag, MIDDLE+double+drag|_translate |translate ||WHEEL |_wheelZoom |zoom ||] ','1|2','','') newCmd('','11.9.9','','*v+12.0;','Ties a specific mouse action to a specific Jmol action.','*1','(mouse action)','(Jmol action)') newCmd('','11.9.9','','*v+12.0;','Ties a specific mouse action to a script defined by the user, replacing its current Jmol action. ','*2','(mouse action)','"script"') newCmd('','13.2','','*v+13.2;','An added "+:" at the beginning of a user action indicates to add this action to the currently bound Jmol action, retaining the Jmol action. This allows creating "hooks" that allow detection of mouse actions.','*3','(mouse action)','"+:script"') newCmd('.break','','','*v+11.4;flow','{#.while~} and {#.for~} loops may be exited using break and truncated using continue. A number following break or continue indicates how many levels of for/while loops beyond the innermost one to break out of or continue.
n = {{*}}.length
for (var i = 1; i <= n; i++) {{:>for (var j = i + 1;j <= n; j++) {{:>> var dis = {{*}}[i].distance({{*}}[j]);:>> if (dis < 1.23) {{:>> print "short i-j: " + i + "," + j + " " + dis%2:>> measure {{atomno=i}} {{atomno=j}}:>> continue;:>> }} else if (dis < 1.77) {{:>> print "medium i-j: " + i + "," + j + " " + dis%2:>> line = "line"+i+"_"+j:>> draw @line {{atomno=i}} {{atomno=j}}:>> break 1;:>> }}:>> print "long";:>}}
}}','0|1','','') newCmd('.continue','','','*v+11.4;flow','See {#.break~}.','0|1','','') newCmd('.bondorder','','','*v+11.4 -- new;bond','Sets the bond order of the selected atoms or bonds. An alternative to the {#.connect~} command.','0|1','','') newCmd('','','','','Sets the bond order to the specified order. Values -1, -1.5, and -2.5 specify HBOND, PARTIALDOUBLE (reverse solid/dash of AROMATIC), and PARTIALTRIPLE2, respectively. ','*1','0.5, 1, 1.5, 2, 2.5, 3, 4, -1, -1.5, -2.5','') newCmd('','','','','Sets the bond order to the specified type. See details at {#.connect~} for PARTIAL N.M .','*2','._connection_options','') newCmd('.boundbox','11.3.44','','*v+11.4 -- expanded capability;set;boundbox','Turns on or off a wire-frame box that contains the model or a designated subset of the model or a designated region of space, and determines the line style and line width (as a decimal number, in Angstroms) of that box. If the atom set is not indicated, the boundbox is drawn around the entire model set (including all models in frames). A decimal number specifies the boundbox line diameter in Angstroms; DOTTED specifies a fine dotted line. For an explanation of center, corners, and vectors, see {#.show~show boundbox}.','0|1','','') newCmd('','','','*v+11.4;','Turns the boundbox on or off around the specified set of atoms. In a multi-model context, if a set of atoms is specified and that set of atoms is within a subset of the models, then the boundbox is only displayed only when the {#.model~} command has made that subset of models displayable. ','*1','._atom_expression{"*"}','._axes_type{"ON"}') newCmd('','','','*v+11.4;','Sets the boundbox to be centered on the atom set or coordinate given as the first parameter and extending by a vector to a corner specified by the second parameter. If the third parameter is not given, no change in the visibility of the boundbox or its line style is made.','*2','._atom_expression_or_coord','._coordxyz','._axes_type{"unchanged"}','') newCmd('','','','*v+11.4;','Sets the boundbox based on two corners, each specified by the center off an atom set or a coordinate. If the fourth parameter is not given, no change in the visibility of the boundbox or its line style is made','*3','CORNERS','._atom_expression_or_coord','._atom_expression_or_coord','._axes_type{"unchanged"}') newCmd('','11.9.12','','*v+12.0;ticks','Sets the parameters for ticks along the first three boundbox edges. The parameters are similar to those of {#.axes~axes TICKS}, but unit cell scaling is not an option.','*41','TICKS X|Y|Z','{{major,minor,subminor}} FORMAT ["%0.2f", ...] ','SCALE {{scaleX, scaleY, scaleZ}} | x.xx','') newCmd('','11.9.19','','*v+12.0;','With any of the above parameters, specifies to scale the boundbox by (a) multiplying by the scaling factor if positive or (b) adding to the size if negative. ','*42','SCALE','x.xx') newCmd('','11.9.19','','*v+12.0;','Sets the current boundbox to match the extent of a given isosurface. ','*5','$isosurfaceID','') newCmd('.cache','13.1.2','','*v+13.2;cache;load','Establishes a memory-based cache of data. A summary of the contents of the cache can be obtained with show cache.','0|1','','') newCmd('','13.1.2','','*v+13.2;cache','Clears the cache','*2','CLEAR','') newCmd('','13.1.2','','*v+13.2;','Adds a file to the memory cache as a set of bytes.','*1','ADD','"filename"') newCmd('','13.1.2','','*v+13.2;','Removes the specified file from the cache. cache REMOVE ALL is the same as cache CLEAR.','*3','REMOVE','"filename"') newCmd('.calculate','','','*v+14.2 -- adds DSSR (Defining the Secondary Structures of RNA);calc','Calculates specific quantities.','0|1','','') newCmd('.calculate','','','*v+13.0 -- adds calculation of partial charges using the MMFF94 charge model;calc','Calculates specific quantities.','0|1','','') newCmd('.calculate','','','*v+12.0 -- adds calculations for standard hydrogen bonds, hydrogens, pointgroup, straightness, DSSP-based structure, and struts;*v-13.0;calc','Calculates specific quantities.','0|1','','') newCmd('','','','*v+11.2;','Calculates alternating single and double aromatic bonds for all bonds of type AROMATIC. If just one bond of a conjugated system is specified as AROMATICSINGLE or AROMATICDOUBLE, then all bonds of that system will be consistent with that bond. . For example:
reset aromatic;
connect (atomno=3) (atomno=4) AROMATICDOUBLE;
calculate aromatic;
or
select carbon and within(1.6, {{0 0 0}});
connect (selected) aromatic modify;
calculate aromatic;
','*10','AROMATIC','') newCmd('','14.4.1','','*v+14.4;','Explicitly calculates (or recalculates) Cahn, Ingold, Prelog chirality, including (R, S, E, Z, M, P, r, s) for the currently selected atom set. Jmol\'s calculation is a nearly complete implementation of the IUPAC recommendations of 2013, only lacking topological chirality (related to chains that pass over or under a rigid system such as a benzene ring). calculate chirality by itself reports a list of chiral designators, providing the values for the .chirality and .ciprule atom properties and %[chirality] and %[ciprule] atom label options, as well as creating _M.CIPInfo, which contains detailed information relating to the calculation for each center. An added optional atom set limits the calculation to a specific set of atoms.','*11','CHIRALITY','._atom_expression') newCmd('','13.3.1','','*v+14.0;','Calculates formal charges based on hybridization and bonding.','*12','FORMALCHARGE','') newCmd('','12.0.RC1','','*v+12.0;','If no atom sets are indicated, performs the same as {#.hbonds~hbonds calculate}. Three types of hydrogen bond calculation are available.
[||RasMol-type pseudo-hbonds (PDB/mmCIF files only)|select not hydrogen; set hbondsRasmol TRUE; calculate HBONDS | The default calculation when no hydrogen atoms are selected. Creates pseudo-hydrogen bonds only between protein amide and carbonyl groups or between nucleic acid base pairs using a RasMol-like (DSSP) calculation. Bonds can be differentiated using {#.color bond object~color hbonds TYPE} (see {http://jmol.sourceforge.net/jscolors/#Hydrogen%20bonds~} or {#.color bond object~color hbonds ENERGY} (red - high energy, less stable; blue - low energy, more stable). This energy is calculated according to the equation given at {http://en.wikipedia.org/wiki/DSSP_%28protein%29~http://en.wikipedia.org/wiki/DSSP_(protein)}||Creates Jmol-type pseudo-hbonds|select not hydrogen; set hbondsRasmol FALSE; calculate HBONDS | pseudo-hydrogen bonds between O or N atoms on proteins and nucleic acid (but not limited to backbone atoms), and other atoms, including water oxygens. Psuedo-hydrogen bonds are not calculated between two water molecules. Uses an algorithm developed for Jmol involving parameters (see below). These hbonds are not colorable by energy.|| Standard hydrogen bonds|all cases where hydrogens are selected followed by calculate HBONDS| Creates bonds from hydrogen atoms on O or N to other O and N atoms, regardless of whether they are in macromolecules. When two atom sets are used, hydrogen atoms must be present in the first set; other atoms in that set will be ignored. Coloring of standard hydrogen bonds by "energy" is possible, but the value associated with that color should be be taken as a true energy. Note that you can add hydrogen atoms to a PDB file using set pdbAddHydrogens prior to loading a PDB file.||] If two atom sets are provided, hydrogen bonds will be between atoms in the first atom set and atoms in the second atom set. These sets may overlap, and a common invocation of the command is simply calculate HBONDS, which uses the currently selected atoms for both sets. When the RasMol calculation is not used, the parameters hbondsAngleMinimum (default value 90) and hbondsDistanceMaximum (default 3.25 for pseudo-hydrogen bonds; 2.5 for standard hydrogen bonds) are used. The angle referred to is the bond angle between the candidate hydrogen (or pseudo-hydrogen) bond and the covalent bonds to the atoms involved. The idea is that all bond angles that include hydrogen bonds should be greater than some designated value. Note that the default value of 90 degrees may be too large for the general case. Values as low as 70 or 80 may be approriate in some cases, and experimentation may be necessary. The distance maximum is used only for pseudo-hydrogen bonds when its value is set larger than 2.5. ','*22','HBONDS','._atom_expression','._atom_expression','') newCmd('','12.0.18','','*v+12.0;','Calculates the hydrogen bonds that define the DSSP structures generated with {#.calculate~calculate STRUCTURE}. These are not all of the "hydrogen bonds" identified by DSSP; rather they are just the ones that define the helixes, sheets, and bridges. ','*23','HBONDS','STRUCTURE') newCmd('','11.9.19','','*v+12.0;;','Adds hydrogens at calculated positions based on bonding patterns at the designated atoms or, if no atoms are specified, at all atoms. Note that this command is not intended to be used for the assignment of hydrogen atoms in standard PDB files. All atoms must have any formal charges already designated, and all multiple bonds must be in place. If you want to add hydogen atoms to a standard PDB structure, use set pdbAddHydrogens prior to loading the file. Jmol will then do a proper hydrogen atom addition, to proteins, nucleic acids, carbohydrates, and all ligands. See {#set_pdbaddhydrogens~set pdbAddHydrogens}, below. Adding TRUE also includes connect aromatic modify; calculate aromatic to add multiple bonding.','*240','HYDROGENS','._atom_expression{"*"}') newCmd('','12.3.24','','*v+11.6;;','Calculates relatively reasonable partial charges using the MMFF94 charge model. Multiple bonding is required. The currently selected atom set is used or, if no atoms are selected, the currently visible atom set.','*241','PARTIALCHARGE','') newCmd('','11.5.52','','*v+11.6;;','Calculates the point group symmetry for a symmetrical or nearly symmetrical molecule. The calculation is carried out only on the currently selected atoms and is limited to at most 100 selected atoms. The symmetry-determining algorithm looks for proper and improper rotation axes using a variety of methods. In each case, a test is made as to whether all atoms subjected to a specific rotation or reflection map onto the positions of some other atom.The extent to which imperfections in symmetry will be tolerated depends upon two adjustable paremeters. pointGroupDistanceTolerance (default 0.2 Angstroms) determines the maximum distance between a rotated atom and another atom in the molecule. pointGroupLinearTolerance (default 8.0 degrees) determines whether a potential axis matches one that has already been discovered. Setting these values to higher numbers allows more flexibility in terms of atom positions, but also may result in molecules being reported with higher symmetry than they really have.','*25','POINTGROUP','') newCmd('','11.3.47','','*v+11.4;','Calculates for each atom the property surfaceDistance, which is the distance of the atom to a "shrink-wrap" surface surrounding the set of preselected atoms (usually the entire model, often without solvent molecules). Values for atoms OUTSIDE this surface are not generally valid due to the nature of the calculation. Use calculate surfacedistance FROM {{atom expression}} instead for calculating distances outside of a van der Waals surface surrounding a subset of atoms in a model. {#.isosurface~isosurface sasurface map property surfaceDistance} then creates an isosurface colored by distance from the specified subset; or either {#.color~color surfaceDistance} or {#.color~color property surfaceDistance} colors the atoms along the same lines.','*9','SURFACEDISTANCE','WITHIN','._atom_expression','') newCmd('','11.3.47','','*v+11.4;','Calculates for each atom the property surfaceDistance, which is the distance of the atom to a van der Waals surface surrounding the specified subset of atoms of the model. {#.isosurface~isosurface sasurface map property surfaceDistance} then creates an isosurface colored by distance from the specified subset; or either {#.color(color scheme)~color surfaceDistance} or {#.color(color scheme)~color property surfaceDistance} colors the atoms along the same lines.','*8','SURFACEDISTANCE','FROM','._atom_expression','') newCmd('','11.5.43','','*v+11.6;','Calculates a value for "straightness" (ranging from -1 to 1) within a biomolecular polymer (protein or nucleic) as defined by the following equation:
straightness = 1 - 2 * (acos(qi / qi-1 * qi+1 / qi) / PI)
where qi is the {#.quaternion~} defining the frame of the ith amino acid or nucleic acid. A value of 1 for straightness indicates that the three amino acids (i-1, i, i+1) are perfect rotations around the same helical axis. The straightness can then be displayed as part of a label using the %T format code or as a color using {#.color~color straightness.','*3','STRAIGHTNESS','') newCmd('','10.9.62','','*v+11.4;','Calculates the secondary structure of proteins or nucleic acids . Options are based on the type keyword. The calculation is performed for all models containing any of the indicated atoms or, if no atoms are specified, the set of currently selected atoms. For these models all cartoons and other biomolecular shapes are turned off. The next cartoons on command will then show a complete set of cartoons. This command also recalculates the polymer chains making up a protein or nucleic acid. The results are affected by any bonding that has changed via the {#.connect~} command. A typical use involves PDB files that load with incomplete bonding (because the author specified only a fraction of the bonds). After loading, one can issue connect to use Jmol\'s autobonding feature, then calculate structure to regenerate the secondary structure.[||DSSP | Calculates the secondary structure of proteins based on Jmol\'s implementation of {http://en.wikipedia.org/wiki/Secondary_structure~DSSP}.|| RAMACHANDRAN | Calculates protein secondary structure using a Ramachandran-angle-based method original to Jmol. The default DSSP setting is to ignore any file-based backbone amide hydrogens, but this can be changed using set dsspCalculateHydrogenAlways FALSE.||DSSR | {http://x3dna.bio.columbia.edu~Defining the (Secondary) Structures of RNA}. The calculation works equally well with DNA. ...TO DO... ||] If no type is given, DSSP is assumed unless the global setting {#set_defaultStructureDSSP~set defaultStructureDSSP} is set FALSE, in which case RAMACHANDRAN is assumed.','*4','STRUCTURE','type','._atom_expression','') newCmd('','11.9.22','','*v+12.0;','Generates {#.struts~}. Three parameters are used (defaults given): set strutSpacing 6 sets the minimum spacing between struts, set strutLengthMaximum 7.0 sets the maximum length that is allowed for a strut, and strutsMultiple when set TRUE allows multiple struts on a given atom. In addition, set strutDefaultRadius 0.3 sets the default radius for struts.','*5','STRUTS','') newCmd('','14.3.15','','*v+14.4;','Sets values for getProperty("ShapeInfo.Polyhedra"), including .smiles, .smarts, .polySmiles, and .pointGroup. Optionally carries out the calculation for only a selected polyhedra with ID or associated with specific atoms.','*242','SYMMETRY','POLYHEDRA','(id or atom expression)','') newCmd('.capture','13.3.6','','*v+13.4;image','[||
 ||click image to animate||]
||click image to animate||] Application and signed applet only. Captures animations and mouse action results as animated GIF files, sets of GIF files, or sets of PNG files. Which of these options is selected depends upon the form of the file name: [||xxxx.gif|Capture to an animated GIF file. By default, capturing will continue until the CAPTURE END command is issued. If the LOOP keyword is not given (see below), then whether the animation loops or not depends upon the value of {#.animation~animation mode}: PALINDROME or LOOP results in a looping GIF (no distinction, as there is no PALINDROME option in GIF animation) or ONCE (no looping). Frame rate is determined by {#.animation~animation fps}, which has a maximum of 50 frames per second. If a DELAY command is given during this capture, then that delay will be recorded in the GIF file. ||xxxx0000.gif|Capture to a stream of GIF files starting with xxxx0001.gif. "0000" is required.||xxxx0000.png|Capture to a stream of PNG files starting with xxxx0001.png. "0000" is not required and will be appended if absent.||]','0|1','','') newCmd('','13.3.6','','*v+13.4;','CAPTURE alone closes the current capture file if open and stops capturing.','*1','','') newCmd('','13.3.6','','*v+13.4;','Starts capturing to the designated file or file set. Captures for the specified number of seconds. If this number is equal to 0 or absent, capturing is continued until CAPTURE END is issued. ','*21','"filename"','(decimal)') newCmd('','13.3.6','','*v+13.4;','Starts capturing to the designated file or file set, creating images with transparent backgrounds. Alternatively, files may be given the extensions "GIFT" or "PNGT" as for the WRITE command. The "T" will be removed.','*21','TRANSPARENT','"filename"','(decimal)','') newCmd('','13.3.6','','*v+13.4;','Starts capturing to the designated animated GIF file, overriding the setting of {#.animation~animation mode}.','*22','"filename"','LOOP','(decimal)','') newCmd('','13.3.6','','*v+13.4;','Creates a looping animated GIF or a file set that rocks around the specified axis (x, y, or z; default y) the specified number of degrees (default 5). Both the axis and degrees are optional. Rate of frame capture is controlled by setting of spinFPS. Set spinFPS to smaller values for fewer frames. Frames are captured through a rocking sequence; CAPTURE END is ignored.','*3','"filename"','ROCK','axis','degrees') newCmd('','13.3.6','','*v+13.4;','Creates a looping animated GIF or a file set that spins around the specified axis (x, y, or z; default y). Rate of frame capture is controlled by setting of spinFPS (default 30). Set spinFPS to smaller values for fewer frames. Frames are captured through a full 360-degree spin; CAPTURE END is ignored. (To capture just a fragment of a full spin, use SPIN ON; CAPTURE ... ; and then issue CAPTURE END when desired.','*4','"filename"','SPIN','axis','') newCmd('','14.29.39','','*v+14.29;','Creates an animated GIF that captures the given sequence of script commands. Any DELAY command is recorded in the GIF file as a frame delay. (14.29.39)','*5','"filename"','SCRIPT','"...script commands..."','') newCmd('','13.3.6','','*v+13.4;','Temporarily turns capturing off; file is still open at this point.','*6','OFF','') newCmd('','13.3.6','','*v+13.4;','Re-starts capturing.','*7','ON','') newCmd('','13.3.6','','*v+13.4;','Stops capture silently; same as CAPTURE by itself.','*8','END','') newCmd('.cartoon','','structure','disp','Cartoons are the classic shapes the are used to depict alpha helices and beta-pleated sheets. A combination of cartoons and {#.rockets~} can be displayed using cartoons along with {#.set_cartoonRockets~}. The {#.set_rocketBarrels~} option removes the arrow heads from cartoon rockets. In addition, {#.set_cartoonLadders~} removes the bases from nucleic acid cartoons but leaves the ladder "rungs". The {#.set_hermitelevel~hermite level} determines the overall cross-section of the helix and sheet renderings. Hermite levels 6 or higher produces a ribbon cross-section in the shape of an ellipse. Use {#.set_ribbonaspectratio~set ribbonAspectRatio 4} rather than the default value of 16 for a more rounded ellipse.','0|1','','') newCmd('','','','','','*11','._on_off{"ON"}','') newCmd('','','','*v+11.6;','Turns cartoon rendering on and all other rendering (including labels) off.','*13','ONLY','') newCmd('','','','','A negative number also implies ONLY.','*2','._cartoon_radius','') newCmd('.case','','','*v+12.0;math;flow','See {#.switch~}.','0|1','','') newCmd('.cd','','','*v+11.8;cd','Changes the default directory or, with no parameters, displays the default directory. This command sets the Jmol parameter defaultDirectory and also, if a local directory, sets the default directory for writing files (application and signed applet). The directory can be local or it can have a URL prefix such as http:// or ftp://, and it may or may not be enclosed in quotes. Note that x = file("") sets x to the full path to the current default directory while x = defaultDirectory sets x to the current default directory as set by the user by the cd command (full path) or simply by setting the defaultDirectory parameter (which may or may not be the full path).','0|1','','') newCmd('','','','*v+11.8;','Displays the default directory.','*1','','') newCmd('','','','*v+11.8;','Resets the default directory to the base directory of the application or applet.','*2','""','') newCmd('','','','*v+11.8;','Changes the default directory to that specified in the directory name. Quotes are optional. The standard notation of two periods indicates "up one level" as in cd ../temp. Note that forward slash, not back slash, should be used to separate directory names along a path.','*3','"directoryName"','') newCmd('','','','*v+11.8;','(signed applet or Jmol application only) Displays a file dialog box allowing for creating and changing directories on the local system.','*4','?','') newCmd('','','','*v+11.8;','Changes the default directory to that specified in the URL directory specified in the {#.set(files and scripts)~loadFormat} variable.','*5','=','') newCmd('.default','','','*v+12.0;math;flow','See {#.switch~}.','0|1','','') newCmd('.center','','','centering','Sets the center of rotation to be the center of the set of atoms defined by the {#.atom expressions~atom expression}. This is calculated as the mean value of the coordinates of the selected atoms along each of the respective axes. If no atoms are selected then the center is set to the center of the bounding box (the default). The three values can be written using braces as a single coordinate, {{x y z}}, if desired.','0|1','','') newCmd('','','','','Recenters the model on the default center.','1','','') newCmd('','','','','Centers the model on the specified atom set. Along with {#.show~show center}, allows for the reading of the coordinated position of a specific atom using, for example, center [CYS]4.O;show center;center. See also {#.centerAt~}','*1','._atom_expression','') newCmd('','','','','Centers the model on a specified model-frame coordinate','*2','._coordxyz','') newCmd('','','','','Centers the model on a specified {#.draw~} object','*3','._draw_object','') newCmd('.centerAt','','','centering','The centerAt command allows centering of the model in one of three different ways: based on an absolute coordinate position, based on an offset relative to the center of the boundbox (the overall application default), or based on an offset relative to the average position of the currently selected atoms. Centering on a specific atom or atom set without first selecting it is also available using the {#.center~} command. If the three numerical values are omitted, they default to 0.0 0.0 0.0. The numbers can be in the form of a coordinate (with braces), {{x y z}}, if desired.','1|2','','') newCmd('','','','','specifies an absolute coordinate for the center, in Angstroms','2','ABSOLUTE','x y z {0.0 0.0 0.0}') newCmd('','','','','relative to the center of the boundbox, which is defined by the minimum and maximum atom center coordinates along each of the cartesian axes','2','BOUNDBOX','x y z {0.0 0.0 0.0}') newCmd('','','','','relative to the average atom position (also known as the "unweighted center of gravity")','1','AVERAGE','x y z {0.0 0.0 0.0}') newCmd('.cgo','14.3.11','','*v+14.4 -- NEW;draw','The CGO (Compiled Graphical Object) command, introduced in Jmol 14.4, allows the creation of relatively simple graphical objects similar to those used in PyMOL. [|| cgo test1a [
BEGIN LINES
VERTEX 0 0 0 VERTEX 2 2 2
VERTEX 2 2 2 VERTEX 3 2 0
END
] | The basic syntax consists of an ID name (test1a here) followed by an array of keywords and data bounded by BEGIN and END statements. In this case we are simply drawing two lines. || cgo test1c [
BEGIN LINE_STRIP
VERTEX 4 0 0
VERTEX 6 2 2
VERTEX 5 2 0
END
] | LINE_STRIP allows drawing of connected lines. || cgo test1b [
BEGIN LINE_LOOP
VERTEX 4 0 0
VERTEX 6 2 2
VERTEX 5 2 0
END
] | LINE_LOOP allows drawing of a polygon. || cgo test2 [
BEGIN POINTS
COLOR 255 0 0
LINE 0 0 3 0 0 3 2 2
COLOR 0 255 0
LINE 0 0 3 2 2 4 2 0
END
] | COLOR acts on the next line. Only the last six numbers are used in the LINE commands. COLOR can also be used between vertices in LINE_STRIP and LINE_LOOP blocks. ||
cgo test2b [
BEGIN LINE_LOOP
DIAMETER 0.3
VERTEX 4 0 0
VERTEX 6 2 2
VERTEX 5 2 0
END
] |The diameter of the lines can be set. ||
cgo test2 [ SCREEN 20
BEGIN LINE_LOOP
VERTEX 10 10
VERTEX 90 10
VERTEX 90 90
VERTEX 10 90
END
] |Screen coordinates can also be used. [0 0] is in the top left corner. The syntax is CGO [SCREEN z ...]. CGO 2D VERTEX records are read as screen coordinates, with depth of z. z > 0 indicates a percent (0.01 far back; 100 front); z < 0 indicates an absolute screen z value as -z. Note that the VERTEX and other CGO point elements are 2D, not 3D. || load $caffeine cgo test1a [ UVMAP @1 @6 @11 0 0 80 80 1 1
BEGIN LINE_LOOP
VERTEX 0 0
VERTEX 80 0
VERTEX 80 80
VERTEX 0 80
END
] | The UVMAP specifies origin and two 3D vectors, two 2D ranges, and two scaling factors: CGO [UVMAP @origin @x @y x0 y0 x1 y1 scaleX scaleY ...] 2D VERTEX records are scaled and mapped to a plane defined by @origin @x @y. Here we have a parallelogram based on atoms 1, 6, and 11. ||
CGO[ PS @{{point(-5,-5,0)}} @{{point(5,-5,0)} @{{point(-5,5,0)}}] data "PS"
%!PS-Adobe-2.0 EPSF-1.2
%%Creator: Bob Hanson (from NBO)
%%Title: nbo orbital slice
%%CreationDate: 1/26/2015 5:36 AM
%%DocumentFonts: Helvetica
%%BoundingBox: 211 300 428 518
%% note: above numbers are from 0.24*881 0.24*1781 0.24*1256 0.24*2156
%%EndComments
%%EndProlog
0.2400 0.2400 scale
newpath
3 setlinewidth
newpath
881 1256 moveto
1781 1256 lineto
1781 2156 lineto
881 2156 lineto
closepath
stroke
1241 1717 moveto
1250 1713 lineto
...
stroke
%%Trailer
showpage
end "PS" | TThe CGO PostScript option has the syntax CGO [ PS @origin @x @y ] data "PS" [primitive encapsulated postscript data] end "PS" It maps 2D EPS data onto a plane defined by an origin point, an x-axis point, and a y-axis point. This makes it somewhat similar to standard UV mapping of textures. It allows 2D data to be superimposed on a model. Only simple PS commands are available for drawing lines. (For example, it doesn not properly implement stroke. Allowed PS commands include moveto, lineto, newpath, closepath, setlinewidth, and scale. It does not implement PS fill, gsave, or grestore). The EPS %%BoundingBox x0 y0 x1 y1 prolog record is used to map [x0 y0] to @origin, [x1 0] to @x, and [0 y1] to @y. It is being used for NBO contour mapping. ||]','1|2','','') newCmd('.color','','','*v+11.4 -- adds fully customizable color schemes;','In general, the color command takes the following syntax:
color [object] [translucent/opaque] [color, property, or color scheme]
Colors can be designated as one of the {http://jmol.sourceforge.net/jscolors/#JavaScript%20colors~[standard JavaScript colors]}, as a red-green-blue triplet in square brackets, [255, 0, 255], as a red-green-blue triplet expressed as a six-digit hexidecimal number in brackets, [xFF00FF], as a triplet expressed as a point in red-green-blue color space -- {{255,0,255}} or fractional {{0.5, 0.5, 1}} . To specifiy a set of atoms to color, you can either select them first -- select *.N?; color green -- or specify them using {#.atomExpressions~atom expression} notation: color {{*.N?}} green. ','1|2','','') newCmd('','','','*v+11.4;','The color command takes several forms, depending upon the type of object being colored: an atom object, a bond object, a chemical element, or a model object. This section of the guide discusses each of these in turn:
- {#.color (atom object)~}
- {#.color (bond object)~}
- {#.color (element)~}
- {#.color (model object)~}
- {#.color (named object)~}
- {#.color (scheme)~}
color atoms TRANSLUCENT orange
color ribbons TRANSLUCENT 0.5 [255, 165, 0]
select oxygen; color opaque.
If neither TRANSLUCENT nor OPAQUE is specified, OPAQUE is assumed. Thus, color atoms red and color atoms OPAQUE red are synonymous. ','TEXT','[translucent/opaque]','') newCmd('','11.1.31','','*v+11.2;','Besides the standard ways of representing a color, Jmol allows coloring based on properties or by one of the known {http://jmol.sourceforge.net/jscolors/~Jmol color schemes}, which include: [||general schemes| "roygb" (default rainbow), "bgyor" (reverse rainbow), "bwr" (blue-white-red), "rwb" (red-white-blue), "low" (red-green), "high" (green-blue), "bw" (black-white), "wb" (white-black), or "friendly" (an accessibility option for color-blind users)||biomolecule schemes | "amino", "shapely", "chain", "structure", "group", "monomer" || property schemes | "formalCharge", "partialCharge", "surfaceDistance", "absoluteTemperature", "relativeTemperature" (or just "temperature")||user-defined schemes|"user" and its reverse, "resu"; see {#.set userColorScheme~user-defined color schemes}.||]The general color schemes can be used with any property. If color RELATIVETEMPERATURE is used, the color range will depend upon the setting of the {#.set rangeSelected~rangeSelected} flag.
You can also color atom-based objects based on user-defined properties read from an external file or standard Jmol atom properties such as partialCharge that Jmol has read from the model file. In the case of Jmol {#atomproperties~atom properties}, use the keyword PROPERTY followed by the name the property, as in color atoms PROPERTY partialCharge. In the case of user-defined properties (created using the {#.data~} command), which always start with "PROPERTY_", simply give the property name: x = load("mydata.txt");data "property_mydata @x";select model=2;color atoms property_mydata. When using either PROPERTY or PROPERTY_xxx, you can set the absolute range of values that will span the full spectrum of colors of the propertyColorScheme you have chosen. Simply add the keyword ABSOLUTE followed by the minimum and maximum values you wish to use for the two ends of the spectrum. So, for example: color atoms property temperature ABSOLUTE 0.0 30.0. ','TEXT','[color schemes and color PROPERTY xxx]','') newCmd('','','','','Many objects inherit both color and opacity from the underlying associated atom (which, themselves "inherit" their color by default from their associated chemical element). For example, by default a bond will inherit the two colors+translucencies of its endpoint atoms. If you simply \'color atoms translucent\', then both the atoms and the bonds will be translucent. But if you \'color bonds opaque\' or \'color bonds red\' and also \'color atoms translucent\' only the atoms will be translucent.
The level of \'translucent\' can be controlled. The algorithm allows for proper mixing of colors for two translucent objects and approximately correct mixing for more than two overlapping translucent objects. ','TEXT','[color inheritance]','') newCmd('','','colorrasmol','','Sets the previously selected atom set to a color based on a color name or value or one of the {http://jmol.sourceforge.net/jscolors/~[Jmol color schemes]}.','*1','._color_name_or_scheme','') newCmd('','','colorrasmol','*v+12.0;','The additional parameter TRANSLUCENT creates a gradient of translucency from transparent to opaque across the color scheme. ','*1','._color_name_or_scheme','TRANSLUCENT') newCmd('.color (atom object)','','','color;latom;','Sets the color of atom-related objects (atoms, backbone, cartoons, dots, halos, labels, meshribbon, polyhedra, rockets, stars, strands, struts , trace, and vibration vectors).','1|2','','') newCmd('','','coloratoms','','Sets the color of atom-related objects based on a previously selected atom set to a specific color, a color scheme, or back to its default color (CPK), or to inherit the color of its associated atom (NONE). In the case of "color atoms", CPK and NONE both revert to the default color. In the case of "color labels", coloring a label to the background color automatically uses the background contrast color, not the actual background color (white or black, depending upon the background color). If it is desired for some reason to color a label the background color, then the label should be colored instead a color very close to the background color but not it exactly. For instance, to color labels black with a black background, use [0,0,1] instead of [0,0,0].','*2','._atom_object','._color_name_or_scheme') newCmd('','13.1.16','','*v+13.2;','Sets the color of the space-fill sphere of an atom without coloring the bonds leading to that atom or other atom-based objects such as stars and polyhedra.','*3','BALLS','._color_name_or_scheme') newCmd('.color (bond object)','','','color;hbondc','Three types of bonds are distinguished by Jmol for coloring purposes: regular bonds, disulfide bonds, and hydrogen bonds. Each can be colored independently, and hydrogen bond colors in proteins can take on a special coloring scheme based on their connectivity.','1|2','','') newCmd('','','','*v+11.4;','Colors selected bonds a specific color or resets them to inherit the color of their associated atoms.','*1','BONDS','._color_or_none_or_CPK') newCmd('','','','*v+11.4;','Colors disulfide bonds a specific color or resets them to inherit their color from their associated atoms.','*2','SSBONDS','._color_or_none_or_CPK') newCmd('','','','*v+11.4;','Colors hydrogen bonds a specific color or resets them to inherit their color from their associated atoms. Additional HBOND color options include TYPE and ENERGY','*3','HBONDS','._color_or_none_or_CPK') newCmd('','','','','Colors hydrogen bonds based on DSSP "energy" (see {#.calculate~calculate HBONDS} for details).','*4','HBONDS','ENERGY') newCmd('','','','','Colors hydrogen bonds specifically in proteins based on how many residues one end is from the other. Note that to get this effect, one must first execute "hbonds ON" and then issue "color hbonds TYPE".'+' The colors assigned are based on the number of redidues between the interacting H atoms. This TENDS to indicate secondary structure, but is not perfect.The correlation between color and offset are as follows:'+'[||Color|Offset||green|-4||cyan|-3||white|+2||magenta|+3 (turns)||red|+4 (alpha-helix)||orange|+5||yellow|other (e.g. beta-pleated sheet)||]','*4','HBONDS','TYPE') newCmd('.color (element)','','','*v+10.2 -- new;color','You can use the \'color\' command to specify customized default colors that are used for elements. {http://jmol.sourceforge.net/jscolors/#Atoms%20(\'CPK%20colors\')~[default Jmol element colors]}
color carbon limegreen
color hydrogen [x32CD32];
These changes are not molecule-specific; they will continue in effect even if new molecules are loaded. However, in a page with multiple applets, each applet will have its own set of element colors.
If you choose to use this feature, you should consider encapsulating your favorite colors into a script and then executing that script as a subroutine. For example:
script LoadMyFavoriteColors.txt;
load foo.xyz;
load bar.xyz;
Note:
- Custom element colors are independent of and are not affected by the currently selected set of atoms.
- To reset custom element colors, use {#.set%20(default%20color%20scheme)~\'set defaultColors Jmol\'} or \'set defaultColors Rasmol\'.
- \'translucent\' or \'opaque\' cannot be specified as part of the element color specification. (You cannot \'color carbon transparent green\', for instance.)
- At this time only elements can be custom colored. There is no support for customizing other color palettes such as those used for protein chains or groups.
Except for the BONDS option, by default the command does not move any atoms and just reports RMSD. A return of "NaN" indicates that the specified atom sets could not be matched because there was not a 1:1 correlation of atoms. The independent options ROTATE and TRANSLATE allow the option to do just rotation, do just center-of-mass translation, or do both. An optional number of seconds can be added and can be 0 to effect no animation. (The default is 1 second.) For example:[||`style="width:500px"> compare {{2.1}} {{1.1}} SUBSET {{*.CA}} | reports RMSD for the comparison of two models based on alpha carbons only. || compare {{2.1}} {{1.1}} ROTATE TRANSLATE 2.0 | Compares the second model to the first and rotates and translates it into superposition with the first model over the course of 2 seconds.||load files "$tyrosine" "$lysergamide";frame *;
compare {{1.1}} {{2.1}} BONDS "c1ccccc1CCN" rotate translate | flexibly fits tyrosine onto lysergamide based on the aromatic ring and C-C-N atoms only, overlaying the structures over 1 second. Note that the required initial internal bond rotations are carried out even if ROTATION and TRANSLATION are not indicated.||]
See also the {#functionsx=fyfunctions~compare()} function. [1] {http://www.opticsinfobase.org/viewmedia.cfm?uri=josaa-4-4-629&seq=0~Berthold K. P. Horn, "Closed-form solution of absolute orientation using unit quaternions" J. Opt. Soc. Amer. A, 1987, Vol. 4, pp. 629-64.} [2] G. R. Kneller, "Superposition of Molecular Structures Using Quaternions" Molecular Simulation, 1991, Vol. 7, pp. 113-119.','1|2','','') newCmd('','11.9.35','','*v+12.0;','Compares the specified subset of atoms of two models, minimizing the RMSD of paired atom coordinates. If the SUBSET option is not specified, the models are matched atom-for-atom based on "SPINE" atoms (*.CA, *.N, *.C for proteins; *.P, *.O3*, *.O5*, *.C3*, *.C4*, *.C5 for nucleic acids). The keyword ATOMS is optional and may be followed by any number of specific pairs of atom sets to be correlated. For example, compare {{2.1}} {{1.1}} SUBSET {{*.CA}} ATOMS {{10-20}} {{50-60}} {{30-40}} {{20-30}} ROTATE TRANSLATE will correlate the positions of alpha carbon atoms in groups 10-20 and 30-40 of model 2.1 with corresponding atoms in groups 50-60 and 20-30 of model 1.1 and move all atoms in model 2.1 in the process. The result of this atom-atom pairing comparison is essentially the same as the PyMol pair_fit command, though easier to implement and using an exact form of the structure-structure correlation rather than an iterative process.','*12','{{model1}} {{model2}}','SUBSET','{{atomSet}} ATOMS [paired atom list]','') newCmd('','11.9.35','','*v+12.0;','Compares the specified subset of atoms of two models, minimizing the RMSD of paired group quaternion orientations. If no other parameters are included, the models are matched group for group based on the current setting of quaternionFrame. The keyword ORIENTATIONS may be followed by any number of specific pairs of atom sets to be correlated. For example, set quaternionFrame "C"; compare {{2.1}} {{1.1}} QUATERNIONS {{10-20 or 30-40}} {{50-60 or 80-90}} ROTATE TRANSLATE will correlate the orientations of alpha carbon atoms in groups 10-20 and 30-40 of model 2.1 with corresponding orientations in groups 50-60 and 80-90 of model 1.1 and move all atoms in model 2.1 in the process. The result of this orientation pairing comparison gives the best fit of orientations and the best translation, but not necessarily the best rotation to fit coordinate positions. The ORIENTATION option has no corresponding PyMol command.','*8','{{model1}} {{model2}}','ORIENTATIONS','[paired atom list]','') newCmd('','11.9.35','','*v+12.0;','The ORIENTATION option allows for explicit comparison of quaternion arrays rather than atom lists. The result is independent of the setting of quaternionFrame. These arrays can be specified in terms of variables. For example: qset1 = quaternion({{10-20:A/1.1}}); qset2 = quaternion({{20-30:D/2.1}}); compare {{2.1}} {{1.1}} ORIENTATIONS @qset2 @qset1.','*9','{{model1}} {{model2}}','ORIENTATIONS','[paired quaternion array list]','') newCmd('','12.0.RC12','','*v+12.0;','This option allows the alignment of two structures based on SMILES or SMARTS atom matching. The basic idea is to use a SMILES (whole molecule) or SMARTS (substructure) description to find the atoms in one structure that correlate one-for-one with atoms in the second structure, then find the rotation and translation that best aligns them. ','*4','{{model1}} {{model2}}','SMARTS or SMILES','"smartsString"','') newCmd('','13.3.5','','*v+14.0;','Compares a set of atom coordinates to a corresponding ordered set of points.','*5','{{atomSet}}','[array of coordinates]') newCmd('','13.3.5','','*v+14.0;','Compares a subset of atom coordinates to a corresponding ordered set of points.','*6','{{atomSet}}','ATOMS','{{subset1}} [coords1] {{subset2}} [coords2]. . .','') newCmd('','13.3.6','','*v+14.2;','The BONDS option does a flexible fit by first constructing a list of matching dihedrals based on the SMARTS (substructure) string, rotating the bonds into position, then doing a COMPARE operation based on the matching atoms. The preliminary rotation is carried out even if ROTATE and TRANSLATE are not present. In the case of compare {{1.1}} {{2.1}} BONDS "c1ccccc1CCN", the initial internal rotation can be done in a prelminary step using the following command: rotate branch @{{compare({{1.1}},{{2.1}},"c1ccccc1CCN","BONDS")}} 0. See {#.rotate_branch~rotate BRANCH} for details.','*7','{{model1}} {{model2}}','BONDS','"smartsString"','') newCmd('','','','*v+12.4;','compare FRAMES is a powerful method that allows the automated alignment of a set of frames such that each model in a sequence is repositioned to have the closest fit to the model before it. This option is particularly useful for visualizing the results of intrinsic reaction coordinate (IRC) calculations, where each model closely resembles the previous model. In the animations of a cyclohexane ring flip, below, on the left we see the animation as it came from the calculation. It is not at all easy to see what is happening here. On the right, we see the same animation after compare FRAMES. Now it is much clearer (especially when you {https://chemapps.stolaf.edu/jmol/jsmol/simple2.htm?load data/cyclflip2.spt~explore it in 3D}) that the ring flip is a smooth transition from chair to half chair to twist boat and back, without having to go through a boat transition state.
[||
 |
| || frame PLAY|compare FRAMES
|| frame PLAY|compare FRAMESframe PLAY||]
All of the options described below are also available for compare FRAMES. A subset of models can also be specified by adding the models to be aligned before the FRAMES keyword. For example: compare {{file=2}} {{1.1}} FRAMES.','*11','FRAMES','') newCmd('.configuration','','','*v+11.0 -- new;','File types PDB, mmCIF, and CIF allow for the designation of certain atoms to be in "alternative locations" or in "disorder groups". This leads to two or more possible structures. While full treatment of this issue is not possible, Jmol can display the different possible "configurations" described in these files for PDB and mmCIF files. This command selects the specified configuration (but does not {#.restrict} or {#.display} it), and also resets all biomolecular shapes (cartoons, traces, etc.) in the currently displayed frame or frames to use the values of that configuration. Hydrogen bonds are also recalculated. The command configuration by itself, with no number, cycles through the possibilities. The command selects atoms that are in the visible frame(s) See also the atom property {#.atomProperties~configuration}, which allows selection of configurations without updating shapes. ','1','[configuration number]','') newCmd('.connect','','','*v+14.2;bond','The connect command allows real-time bond manipulation, allowing the user or application to connect and disconnect specific atom sets. The general syntax is as follows:
connect [minimum and maximum distances] [source and target atom sets] [bond type] [radius option] [color option] [modify/create option]
(connect by itself deletes all bonds and then creates bonds based on Jmol default bond-generating algorithms, all as single bonds, without respect to what bonding patterns might have been indicated in the model file.)','0|2','','') newCmd('','','','','Distances are given in Angstroms, either as decimals or integers. If only one distance parameter is given, it represents a maximum distance. If neither the minimum nor the maximum distance parameter is given, all connections between the two atom sets are made, regardless of distance. The option to connect atoms based on ranges of percentage of bonding/ionic radii instead of fixed values can be specified using a % sign after a distance.','TEXT','[minimum and maximum distances]','') newCmd('','','','','The source and target atom sets are embedded {#.atom expressions~atom expressions} and therefore must be enclosed in parentheses. If the source atom set is not given, it is taken to be the currently selected atom set, "(selected)". If neither atom set is given, "(selected) (selected)" is assumed. The atom expression "_1" in the second selection set signifies "the atom selected in the first set". Thus, it is possible to select something like (_N) (_O and not within(chain, _1)) -- meaning "all nitrogens and all oxygens not in the same chain as the selected nitrogen." Of course, this would be used with a distance qualifier.','TEXT','[source and target atom sets]','') newCmd('','','','hbond','Unless otherwise specified, connections are automatically introduced as single bonds. Any one of the following bond types may be specified: SINGLE, DOUBLE, TRIPLE, QUADRUPLE, QUINTUPLE, SEXTUPLE, AROMATIC, PARTIAL, PARTIALDOUBLE, HBOND, STRUT or UNSPECIFIED. In appearance, AROMATIC and PARTIALDOUBLE are identical except for which side of the bond is represented by a dashed line. PARTIAL and HBOND are both dashed, but they have different patterns, and newly created hydrogen bonds are only thin lines. Additional bond types include ATROPISOMER (the single bond between two aromatic rings with restricted rotation), AROMATICSINGLE, AROMATICDOUBLE, partial numeric bond order (including -1 for PARTIAL, 1.5 for AROMATIC, -1.5 for PARTIALDOUBLE, 2.5 for PARTIALTRIPLE, and -2.5 for PARTIALTRIPLE2), as well as a fully generalized set of partial bonds indicated with
connect ... partial N.M
where N is the number of lines (from 1 to 5) and M is a binary mask indicating which lines are dashed:
[||M|binary|meaning||1|00001|first line dashed||2|00010|second line dashed||3|00011|first and second lines dashed||4|00100|third line dashed||...|...|...||31|11111|all lines dashed||]
So, for example, we have:
[||partial 1.0|single||partial 1.1|same as "partial"||partial 2.0|double||partial 2.1|same appearance as "aromatic", though not "aromatic"||partial 2.2|partialDouble||partial 3.0|triple||partial 3.1|partialTriple||partial 3.4|parialTriple2||]','TEXT','[bond type]','') newCmd('','','','*v+11.2;','Addition of the keyword "radius" followed by a distance in angstroms allows definition of the radius of a modified or newly created bond. If the modify/create option is absent, then "modify" is assumed; if the bond type is absent, then bonds of any type are set, but their bond type is not changed.','TEXT','[radius option]','') newCmd('','','','*v+11.2;','Addition of a color name or designation optionally along with the keyword "translucent" or "opaque" allows definition of the color and/or translucency of a modified or newly created bond. If the modify/create option is absent, then "modify" is assumed; if the bond type is absent, then bonds of any type are set.','TEXT','[color option]','') newCmd('','','','*v+11.2;','Five additional mutually exclusive options relate to what sort of connections are made. The default when a radius or color option is present is "Modify"; otherwise the default is "ModifyOrCreate". These include:
[||Auto| Adjusts bonding using the Jmol autobonding algorithms, specifically applicable to bond types AROMATIC or HYDROGEN, or when no bond type is given. ||Create | Only new bonds will be created. If a bond of any type already exists between two matching atoms, it will not be affected.||Modify | Only pre-existing bonds will be modified. No new bonds will be created. ||ModifyOrCreate | If the connection fits the parameters, it will be made. Bonds already present between these atoms will be replaced.|| Delete | Delete the specified connections.||]','TEXT','[modify/create option]','') newCmd('','','','','Connect the currently selected atoms with single bonds using autobonding.','*1','AUTO','') newCmd('','','','','Add double and triple bonds to a single-bonded selected atom set. Provided necessary hydrogen atoms are present, the algorithm generally adds double and triple bonds where appropriate (aromatic or otherwise). The algorithm does not test for cumulenes.','*2','AROMATIC','AUTO') newCmd('','','','','Adds hydrogen bonds to the currently selected atom set. ','*3','HBONDS','AUTO') newCmd('','','','','Connect atoms in the currently visible model using a resonance structure configuration found in an NBO .46 or .nbo file, where nbotype is one of alpha, beta, 46, 46a, 46b, nrtstr_n, nrtstra_n, rs_n, rsa_n, or rsb_n.','*4','NBO','nbotype') newCmd('.console','','','*v+11.0 new command;','Throws up a console window from which a user can enter script commands and monitor the messages returned by Jmol as, for example, from the {#.show~} or {#.getProperty~} command. JSmol note: <div id=JmolApplet0_console></div> sets a place on the page for the JSmol console.','0','','') newCmd('.contact','12.2.0','','*v+12.2 -- new;','The contact command is a powerful command capable of displaying a wide range of contact surfaces in stunning detail relating to van der Waals interactions, hydrogen bonds, and clashes. The general format of the contact command is
contact ID "id" {{atomSet1}} {{atomSet2}} [contactOptions] [colorOption]
The first atom set is the "target" atom set, typically a ligand or a subset of a protein. The target atom set is required. Its Van der Waals radius will be used. The second atom set is the interacting atom set and is optional, defaulting to "all other atoms". For example: contact {{ligand}} or contact {{ligand}} {{oxygen or nitrogen}}. The interacting atom set Van der Waals radius can be adjusted using absolute (1.2), percentage (120%), or Angstrom offset from 100% (+1.2) relative to the Van der Waals radius of atomSet2. Several contact options are available and are listed below. Color options are similar to those available for {#.isosurface~isosurfaces}. The color density option produces a result roughly similar to {http://kinemage.biochem.duke.edu/software/probe.php~Kinemage Probe}. Examples can be found at {http://stolaf.edu/people/hansonr/jmol/contact/gordon.htm~} and {http://stolaf.edu/people/hansonr/jmol/contact/ligand.htm~}. [|| |
 |
| |
| ||]
||]contact options
[|| hbond, vdw, clash | just hydrogen bonds, van der Waals near-misses, or van der Waals overlaps that are not hydrogen bonds (clashes). || NCI | {http://pubs.acs.org/doi/abs/10.1021/ct100641a~NCIPlot} promolecular calculation results.|| surface | van der Waals surface around the atom set colored by contact proximity. || cap | similar to surface, but with clashing sections capped off. (Specifically designed for prototyping.) || trim | similar to surface, but trimmed to only display van der Waals radius overlaps. || full | same as trim but with both overlapping van der Waals surfaces displayed. || plane (default) | approximately planar circles of the voronoi surface between the groups trimmed to display only the desired area. Contacts are only generated for atoms in different molecules or more than three bonds apart.|| connect | cigar-shaped objects that connect one interacting atom with another. ||]','0','','') newCmd('.data','','','*v+11.6 adds new capabilities;vdw','The data command allows data to be introduced in-line or via a variable. The command consists of two statements, data "label" and end "label", between which the data are presented on as many lines as desired. "label" may be any string, though strings starting with "property_" are special (see below). Quotes must be used in both the data line and the end line. The first word of the label defines the data type, but the label itself may be any number of words. If the data type is "model" as in the following example, then the data is interpreted as an in-line model (and loaded using the default lattice, if crystallographic). If the data type is "append", then the data is interpreted as a model, and the model is appended either into a new frame or into the last existing frame, based on the setting of set appendNew. Additional data types may be loaded and later {#.show~shown}, but they will be ignored by Jmol.[||background red;
data "model example"
2
testing
C 1 1 1
O 2 2 2
end "model example";show data||]
Note that the data statement itself should not include a semicolon at the end. In the specific case of a model file, if it is desired to use no new-line characters, you can start the data with ¦ (vertical bar) and then use a vertical bar to separate all lines: data "model example"¦2¦testing¦C 1 1 1¦O 2 2 2¦end "model example";show data. For this option you MUST start the data with a vertical bar immediately following the quotation mark closing the label or on the very next line. If the first character is a vertical bar or a new-line character, it is not part of the model. To include data representing more than one file, first define a data separator using set dataSeparator, for example, set dataSeparator "@TILDE@TILDE@TILDE". Then use that separator between data sets. The data command thus provides an alternative to the JavaScript Jmol.js (applet-only) loadInline() function. It can be included in any {#.script~}, and commands can come before and after it for further processing. Note that model data in the system clipboard can also be pasted into the applet console or endered into the application using Edit...Paste for direct introduction into Jmol. Using {#.load DATA~load data} instead of just data you can load model data with all of the loading options of the standard LOAD command. See also {#.show~show data}, {#.getProperty~getProperty data}, and {#.load~load "@x"}. ','1|2','','') newCmd('','','','*v+11.6;props','Jmol 12 Note: If you just want to assign a set of properties to a set of atoms, and you have the properties in a file as a list of numbers, you do not need the DATA command. Simply load the values straight into an atom property directly from the file:
{{*}}.property_xxx = load("myfile.dat")
Atom property data may be loaded into Jmol using the DATA command. In addition, when reading some model file types (CRYSTAL, specifically) Jmol will create property_xxx data automatically -- check the Java console after file loading to see a report of what properties were assigned. To assign properties using the DATA command, first select the atoms to which data are to be assigned. Then, to assign the data, use DATA "property_xxx"...end "property_xxx", where "xxx" is any alphanumeric string. Properties are assigned sequentially to the currently selected atom set, and will be assigned to atoms in the selection set one after another until either the data or the atom list is exhausted. If the data are exhausted before the selected atoms, then the value 0 is recorded for each of the remaining selected atoms. In this way, if only a few atoms need data, only a few can be selected and assigned values. Properties can be checked using atom labels: (label %[property_xxx]). The following special values for xxx read data into Jmol exactly as though read from a model file: atomName, atomType, coord, element, formalCharge, occupancy (0 - 100), partialCharge, temperature, valence, vanDerWaals, and vibrationVector. Any other label will be saved simply under its property name.
To read selected data that is in tab- or space-separated format (for example, from a spreadsheet), specify which column the data is in using set propertyDataField = n, (n = 1 being the first column), prior to the data command. Lines having fewer than n tokens will be ignored. If the data is fixed-column format, then n is the starting column number, and set propertyDataColumnCount to be the number of columns to assign to this field. Lines shorter than the required number of characters will be ignored. Setting propertyDataField to 0 indicates that no data field is present, and data are to be read sequentially.
Atom selection need not be contiguous. If you want to associate specific data with specific atom numbers, a column containing these atom numbers (starting with 1 for each model loaded) can be specified using propertyAtomNumberField = m. If the data is fixed-column format, then m is the starting column number, and also set propertyAtomColumnCount to be the number of columns to assign to this field. Specifying 0 for m indicates that the set of currently selected atoms should be assigned values from the data.
Atom property data may also be loaded from variables. To do this, use add "@x", where x is a variable name within the quotation marks defining the first parameter of the data command: DATA "property_partialCharge @x" or DATA "property_mydata 66 3 @x". No end line is required. The variable x should already contain a list of numbers, perhaps from using the x = {#.functions~load}("myfile.dat"); perhaps just by creating a string of numbers: x = "2.3 3.4 5.6 7.8...".
The {#.functions~data()} function also allows direct conversion of data into arrays, which can be directly stored in an atom property using, for example,
{{*.CA/2.1}}.temperature = data(load("mydata.dat"),6,0,3)
meaning, "Read the file mydata.dat and store the sixth free-format field of each line starting from the third line as the temperature of the C-alpha carbons of model 2.1." ','TEXT','Setting atom properties','') newCmd('','','','*v+11.0;','Defines a set of data in line, ending with a matching end "label", where "label" is any string. Quotes are required. Certain labels have special meaning and are described more fully below.','*11','"label"','') newCmd('','','','*v+11.0;','Clears the data table.','*2','CLEAR','') newCmd('','','','*v+11.0;','Defines the USER set of van der Waals radii on an element-by element basis. Entries may be separated by semicolons or entered one per line. . In the example given here, carbon (atomic number 6) is given a radius of 1.7, and nitrogen (atomic number 7) is given a radius of 1.8. ','*3','"element_vdw" 6 1.7; 7 1.8 END "element_vdw"','') newCmd('','','','*v+11.6;','Defines a set of data with the given label from a variable (variable x in this case). Quotes are required, but no end command is required.','*12','"label @x"','') newCmd('','','','*v+11.6;','Defines a data set to be paired x,y data inline and ending with end data2d_xxx, where xxx can be any alphanumeric string.','*13','"data2d_xxx"','') newCmd('','','','*v+11.6;','Defines an atom property based on data provided next inline and ending with end property_xxx, where xxx can be any alphanumeric string. propertyAtomField, propertyAtomColumnCount, propertyDataField, and propertyDataColumnCount override set values for these parameters.','*16','"property_xxx propertyAtomField propertyAtomColumnCount propertyDataField propertyDataColumnCount"','') newCmd('','','','*v+11.6;','Defines an atom property based on data provided next inline and ending with end property_xxx, where xxx can be any alphanumeric string. propertyAtomField and propertyDataField are optional, and if provided override the set values for propertyAtomField and propertyDataField.','*15','"property_xxx propertyAtomField propertyDataField"','') newCmd('.define','','','*v+11.2 -- new DYNAMIC_ prefix;lload;rreset','Defines the specified atom selection variable to be the atoms selected by the {#.atom expressions~atom expression}. Generally, they are calculated once. However, if "DYNAMIC_" is at the beginning of the variable name, as, for example, "DYNAMIC_myatoms", then whenever that variable is used it is recalculated. (When used, the prefix "DYNAMIC_" should be dropped.)
IMPORTANT NOTE: The DEFINE command should be used with some discretion. If you should define a term that later becomes a reserved keyword to any command in any future version of Jmol, your page may not be compatible with that future version. A simple way to avoid this situation is to put a tilde (@TILDE) in front of any definition you make. Do not use a leading underscore (_), as there are many "hidden" reserved definitions that start with that character.
Dynamic or not, definitions hold only until the next {#.initialize~}, {#.load~}, or {#.zap~} command is issued.
This command is part of the legacy command set of Jmol. The "variables" produced are only for atom selection. They are not {#.jmolmathjmolvariables~[Jmol Math]} variables. Consider using Jmol Math variables instead. For example, aa = {{TYR}} instead of DEFINE aa TYR. These definitions are static and persistent until a {#.reset~reset VARIABLES} or {#.initialize~} command is issued.','2','._variablename','._atom_expression') newCmd('.delay','','','pause','Causes the screen to refresh and the script to stop executing for the specified number of seconds. See also {#.set_delaymaximumms~set delayMaximumMs}. Note that the delay command is skipped when executed within a function that is returning a value to a variable, as in
:>>function d() {{ delay 1 }}:>>print d()
','1','.on','') newCmd('','','','','','1','._time_delay','') newCmd('.delete','11.9.19','','calc;cache','The DELETE command deletes atoms or identified objects. DELETE by itself deletes all atoms, similarly to the way select by itself selects all atoms.','1|2','','') newCmd('','','','','Deletes the specified atoms. For example: DELETE hydrogen or DELETE atomno > 30 . Note that delete by itself deletes all atoms, similarly to the way select by itself selects all atoms.','2','._atom_expression','') newCmd('','','','','Deletes the specified objects (from {#.draw~}, {#.isosurface~}, etc. using the ID parameter). Wildcards may used. For example, delete $d* will delete all identified objects starting with the letter d. delete $* deletes all such objects.','3','$','[object id]') newCmd('','14.1.11','','','Deletes the specified information saved with the {#.save~} command, such as "Orientation_o1". All information matching either at the beginning (delete $Orientation_) or the general name (delete $o1) will be deleted from memory. ','4','$','[save id]') newCmd('.depth','11.1.5','','slabdepth',' Slab and Depth together control the percentage of the molecule to be displayed based on clipping planes. All of the commands available for {#.slab~} are also available for {#.depth~}.','1|2','','') newCmd('.dipole','','','*v+11.0 -- NEW;','The dipole command allows for the drawing of a bond or molecular dipole arrow with or without a cross near the tail. Note that without the cross, since it can be drawn from any point in molecular space to any other, a "dipole" can be used by a web page developer for a simple arrow in order point to some particular aspect of the model having nothing to do whatsoever with dipole moments.The values of each dipole can be set by the user or will be estimated using partial charge data or molecular dipole information if available in the loaded model file. Only a very crude calculation is used to estimate at least the direction of all bond dipoles. The general syntax of the dipole command is as follows:
dipole [objectID] [modifying parameters] [positions]
','0|1','','') newCmd('','','','*v+11.6;','The optional identifier such as "bond1" that can be referred to in later scripts as $bond1. These words are arbitrary and if preceded by the optional keyword ID may be any string. The special identifier BONDS (without the keyword ID) refers to the entire collection of bond dipoles -- those dipoled defined specifically as between two atoms. Similarly, the special identifier MOLECULAR (without the keyword ID) is primarily for files for which molecular dipole information is available. The value and placement of this dipole can also be set using the dipole command.','TEXT','ID [object id]','') newCmd('','14.1.16','','*v+14.2;','Defines a dipole based on the given subset of atoms.','*2','ID [object id]','ALL','._atom_expression','') newCmd('','','','','The dipole is defined using a small set of parameters. These include:
[||CROSS
NOCROSS| include (default) or do not include a 3D cross near the tail of the arrow. ||DELETE|Deletes the specified dipole if an identifier is given or all dipoles if no identifier is given; not used with any other parameters.||WIDTH x.xx|The width of the dipole in Angstroms. The default value is 0.005 Angstroms.||OFFSET x.xx
OFFSET n | Dipoles are by default centered between the two endpoints. The OFFSET value sets the offset of the dipole from this position along the axis of its endpoints. In Angstroms if a decimal number is given; in percent of the distance between the two endpoints if an integer is used. ||OFFSETSIDE x.xx|The offset of the dipole in Angstroms perpendicular to the axis of its endpoints. The default value is 0.4 Angstroms for atom-based dipoles and 0 for the molecular dipole.||VALUE x.xxx | A decimal number indicates the value of the dipole. Overall scaling is accomplished either by setting this number or using {#.set (misc)~set dipoleScale}. The VALUE keyword is optional.||]','TEXT','[modifying parameters]','') newCmd('','','','','The positions of the endpoints of the dipole are set either using embedded atom expressions in parentheses, such as (atomno=1), or using a specific point in molecular space, {{x y z}}, either as a cartesian coordinate or a {#.fractional coordinates~fractional unit cell coordinate}. If two atoms are designated, then the dipole becomes a member of the "bonds" dipole collection and can be colored with that group. If a set of atoms is indicated, the geometric center is used. Thus, (*) indicates the geometric center of the molecule.','TEXT','[positions]','') newCmd('.display','','','*v+12.2 -- adds ADD, REMOVE, and GROUP keywords;dhide','The opposite of {#.hide~}. Display is similar to {#.restrict~} in its action, but it is far more flexible. Atoms can be added to the displayed set using display displayed or ... or removed from the hidden set using display displayed and not .... Unlike restrict, the display command does not delete the hidden shapes. Thus, after restrict none all cartoons, traces, spacefill spheres, and bond sticks are effectively deleted. In contrast, display none is easily reversed using display all. or deselected with select not hidden. Atoms can be added to the hidden set using hide hidden or ... or removed from the hidden set using hide hidden and not ..... In addition, bonds can be hidden or displayed.','0|1','','') newCmd('.display','','','*v-12.2;dhide','The opposite of {#.hide~}. Display is similar to {#.restrict~} in its action, but it is far more flexible. Atoms can be added to the displayed set using display displayed or ... or removed from the hidden set using display displayed and not .... Unlike restrict, the display command does not delete the hidden shapes. Thus, after restrict none all cartoons, traces, spacefill spheres, and bond sticks are effectively deleted. In contrast, display none is easily reversed using display all. or deselected with select not hidden. Atoms can be added to the hidden set using hide hidden or ... or removed from the hidden set using hide hidden and not ..... In addition, bonds can be hidden or displayed.','0|1','','') newCmd('','','','','Displays a set of atoms. Displays atoms and associated structures (bonds, halos, stars, cartoons, etc.) and hides all others. The optional parameters ADD and REMOVE prior to the atom expression allow quick alternatives to "displayed or..." and "displayed and not...". ','*10','._atom_expression','') newCmd('','','','','Adds a set of atoms to the displayed set. Equivalent to "DISPLAY displayed or (...)".','*11','ADD','._atom_expression') newCmd('','','','','Removes a set of atoms from the displayed set. Equivalent to "DISPLAY displayed and not (...)".','*12','REMOVE','._atom_expression') newCmd('','','','','Displays a set of bonds using bondset notation. For example, display [{{3:4 5:9 10 13}}]. See {#jmolmathjmolmathvariabletypes~Jmol Math Variable Types}.','*3','[{{...}}]','') newCmd('','','','','Displays all bonds.','*4','BONDS','') newCmd('.dots','','','disp','Creates a dotted surface around atoms. DOTS ON, DOTS OFF, and DOTS ONLY act on the currently selected atoms; all others add the currently selected atoms to the atoms with dots and then apply their settings to all atoms with dots. See also the {#.geoSurface~} command and the settings {#.set_dotdensity~set dotDensity} and {#.set_dotscale~set dotScale}, {#.set_dotsselectedonly~set dotsSelectedOnly}, and {#.set_dotsurface~set dotSurface}.','0|1','','') newCmd('','','','','Turns dots on or off for the currently selected atoms.','*11','._on_off{"ON"}','') newCmd('','','','*v+11.6;','Turns dot rendering on and all other rendering (including labels) off.','*13','ONLY','') newCmd('','','','','Draws dots at the van der Waals radius. See {misc/radii.xls~radii.xls}.','*2','VANDERWAALS','') newCmd('','','','','Draws dots at the (nominal) ionic radius if the atom\'s formal charge has been read and is nonzero; uses the nominal covalent bonding radius otherwise ({misc/radii.xls~radii.xls}).','*3','IONIC','') newCmd('','','','*v+11.0;','Draws dots at the indicated percent of the van der Waals radius for each atom (maximum value 1000%).','*4','nn%','') newCmd('','','','*v+11.0;','Draws dots at the indicated radius in Angstroms for each atom (maximum value 10.0 Angstroms). A negative number also implies ONLY.','*5','(decimal)','') newCmd('','','','','Draws dots at the indicated distance in Angstroms beyond the van der Waals radius for each atom (maximum value 10.0 Angstroms). The "+" sign is required.','*6','+(decimal)','') newCmd('','','','*v+11.6;','Draws dots at the radius corresponding to the minimum anisotropic displacement parameter for the selected atoms factored by the given percentage. See also {#.ellipsoid~}.','*7','ADPMIN','n%') newCmd('','','','*v+11.6;','Draws dots at the radius corresponding to the maximum anisotropic displacement parameter for the selected atoms factored by the given percentage. See also {#.ellipsoid~}.','*8','ADPMAX','n%') newCmd('.draw','','','*v+11.6 Jmol 11.6 adds ARROW ARC, ARC, CIRCLE, and ID options, allows for mixed-type point sets, and special options Ramachandran, Quaternion, and PointGroup;iso, plot','The draw command allows for the insertion of points, lines, and planes to a model. The general syntax of the draw command is as follows:
draw [objectID] [modifying parameters] [positions]
','0|1','','') newCmd('','','','*v+11.6;','The optional identifier such as "line1" or "plane2" that can be referred to in later scripts as $line1 or $plane2. These words are arbitrary and if preceded by the optional keyword ID may be any string. Specifically for the options ON, OFF, and DELETE, the id may include a wildcard character (asterisk). Thus, draw v* off turns off all drawn objects having an ID starting with the letter "v".','TEXT','ID [object id]','') newCmd('','','','*v+11.0;','Several options allow for modifications of some or all of the object types described below. These include:[||COLOR (color)|Sets the color of the drawn object at the time it is created. (The {#.color (model object)~color} command can be used retroactively as well.) ||CROSSED|Two lines (already drawn objects) specified next are crossed; switch the order of vertices for defining a plane.||DELETE|Deletes the object if an identifier is given or all drawn objects if none is given; not used with any other parameters. "*" can be used as a wild card either at the beginning or end of the identifier. For example, DRAW pt* DELETE.||DIAMETER n|Sets the number of pixels for the diameter of points, lines, arrows, and curves. Note that this means that zooming of the model does not change the width of these objects. n may be a decimal value x.x, same as WIDTH x.x.||DIAMETER n [x y %]|Draws a sphere in the 2D window (in front of atoms) of size n x% from the left and y% from the bottom of the screen. For example, to create a key of elements and their colors:
:> background white :> load $caffeine :> function createElementKey() {{ :>> var y = 90 :>> var fontSize = _height*20/400 :>> for (var e in {{*}}.element.pivot){{ :>> var c = {{element=@e}}.color :>> draw ID @{{"d_"+ e}} diameter 2 [90 @y %] color @c :>> set echo ID @{{"e_" + e}} [91 @{{y-2}} %] :>> echo @e :>> font echo @fontSize bold sansserif :>> color echo black :>> y -= 5 :>> }} :> }} :> createElementKey()
||DIAMETER n [x y]|Draws a sphere in the 2D window (in front of atoms) of size n x pixels from the left and y pixels from the bottom of the screen.||FIXED/MODELBASED|Sets whether the surface generated is to be associated with the fixed window -- and thus appear with all frames/models -- or is to be associated with the currently displayed model (the default).||LIST|Lists all DRAW objects. Not used with any other parameters.||ON/OFF|Turns on or off the identified object or all drawn objects if no identifier is given; not used with any other parameters.||PERP
PERPENDICULAR|Draw this object perpendicular to the next indicated object.||REVERSE|Reverse the order of vertices used if the next object listed is a line.||ROTATE45|Rotate a perpendicular plane to a line by 45 degrees.||LENGTH (decimal)|The length for a line/axis in Angstroms. The keyword LENGTH is optional.||OFFSET {{x y z}}| offsets the object by the given x, y, and z distances.||SCALE (decimal)
SCALE (integer)|SCALE with a decimal value indicates a scaling factor for the drawn object. For example, draw SCALE 1.5 (atomno=1) (atomno=2) draws a line with length 1.5 times the distance from atom 1 to atom 2. The line is centered on the two atoms. The keyword SCALE is required in this case. Note that a draw command can consist of just an identifier and a scale. Thus, if $line1 is already defined, draw line1 SCALE 1.3 will rescale that line to 130% of the distance between its defining points. SCALE with an integer number indicates a percent scale. The keyword SCALE is optional in this case.||VERTICES| Generally the geometric center of an atom expression or drawn object is used for positioning. Added just before the atom set or object name reference, VERTICES indicates to use all vertices, not just the center point of the atoms in the expression or the points in the object.||WIDTH x.x|Sets the diameter of points, lines, arrows, and curves to a given width in Angstroms. Objects drawn will be scaled based on perspective and zoom setting.||"text"| A simple text label can accompany drawn objects. The text appears near the first point. Text starting with ">" will be associated with the last point instead of the first. The ">" character will be stripped before the text is displayed. If set drawHover TRUE has been issued, this text will appear only when the object is hovered over.||] ','TEXT','[modifying parameters]','') newCmd('','','','*v+11.0;','Positions define position of the point, the endpoints of the line/axis, or the corners of a plane. Positions can be indicated in any combination of any of the following four ways. Mixed types are processed in the order given on the command line.
[||{{x, y, z}}|a model-frame cartesian coordinate, in braces, ||{{x, y, z/}}|for crystal structures, a unit-cell {#.fractional coordinates~fractional coordinate}, in braces, ||$object|a previously defined drawing object such as $line1 or $plane2, preceded by "$". ||(atom expression)|an atom expression, in parentheses.||@{{ {{atomExpression}}.split()}} | atom expressions split based on model index .||] In addition, if two parameters are given, where the first evaluates to a point and the second is a four-vector {{a b c d}}, a line is drawn from the point to the nearest point on the plane defined by ax + by + cz + d = 0. (Note that because quaternions and axisAngle are stored in quaternion format, which is internally the same as that used for planes, if a quaternion or axisAngle is used in this context, the line will be along the axis of rotation represented by the quaternion or axisAngle q to a point on a plane defined by d = q.w = cos(theta/2), where theta is the rotation angle).','TEXT','[positions]','') newCmd('','','','*v+11.0;','Display options for DRAW are indicated in the following table. These may be given at the end of the definition or in a later command having just these keywords and the identifier (or "ALL") of the desired draw object.
[||FILL/NOFILL| Display the drawn opject as a smoothly filled surface.||FRONTONLY/NOTFRONTONLY| Display only the front half of the surface. This can be useful when the options mesh nofill are used. ||FRONTLIT /BACKLIT /FULLYLIT| In some cases the object may appear flat. Using BACKLIT will fix this; FULLYLIT makes exterior and interior surfaces bright; FRONTLIT is the default setting. ||MESH/NOMESH| Display a mesh of lines intersecting at the vertexes used to construct the object. For cylinders, the combination MESH NOFILL creates a cylinder with no end caps. ||ON/OFF| Turn the object ON or OFF, but do not delete it. ||OPAQUE/TRANSLUCENT n| Display the object as an opaque or translucent object. Several translucent options are available; see the {#.color~} command. ||]','TEXT','[display options]','') newCmd('','','','','Draws a theta-degree arc in a plane perpendicular to the line pt1--pt2 starting at nDegreesOffset degrees rotation from reference point {{ptRef}} at a point fractionalOffset from pt1 to pt2. The SCALE parameter is used to set the diameter of the overall circle containing the arc; the DIAMETER parameter sets the diameter of the curved arc line itself. ARROW ARC adds an arrow head.','*10','ARC','{{pt1}} {{pt2}} {{ptRef}} {{nDegreesOffset theta fractionalOffset}} ') newCmd('','','','','As above, but uses the plane as a reference to define a perpendicular axis. ','*11','ARC {{pt1}} {{plane}} {{ptRef}} {{nDegreesOffset theta fractionalOffset}}','') newCmd('','','','','Draws a straight (two-point) or curve (more than two-point) arrow. The keyword BARB indicates "fish-hook" half-arrows as for organic chemistry mechanisms involving radicals. ','*12','ARROW','{{pt1}} {{pt2}} {{pt3}} ...') newCmd('','','','*v+14.0;','Draws an ARROW based on an array of points. ','*13','ARROW','[array of points]') newCmd('','12.1.48','','*v+12.2;','The DRAW command can be used to draw organic "mechanistic arrows." You need to specify two atoms, two bonds, an atom and a bond, or a bond and an atom. Atoms are identified using the keyword ATOM followed by an atom expression; bonds are identified using the keyword BOND followed by two atom expressions. For example: draw ID "arrow1" ARROW ATOM @1 BOND @1 @2, which draws a curved arrow from atom 1 (atomno=1) to the middle of the bond connecting that atom to atom 2. These drawn objects are then "connected" to the atoms, so if the atoms are not displayed, the arrow is not displayed either. Of course, individual arrows can be hidden independently of their associated atoms as well. ','*20','ARROW','ATOM/BOND') newCmd('','11.9.17','','*v+11.0;','Draws the currently defined {#.boundbox~}. Note that by default this is a solid. To show just edges, use options MESH NOFILL.','*221','BOUNDBOX','') newCmd('','14.21.2','','*v+14.22;','Draws the (approximately) best box around the selected set of atoms, not necessarily oriented by the x,y,z axes. Note that by default this is a solid. To show just edges, use options MESH NOFILL.','*222','BOUNDBOX','BEST') newCmd('','','','','Draws a circle with center at pt1 (which may be an atom expression) in the plane perpendicular to the line between pt1 and pt2. If pt2 is not specified, the circle appears in 2D and remains in the plane of the screen when the model is manipulated. Together, the SCALE and DIAMETER parameters set the overall size of the circle. If no DIAMETER is given and an atom expression is given for pt1, the default diameter is one that includes those atoms; otherwise the default diameter is 1.0 Angstrom. The circle will be filled to form a solid disk unless the MESH NOFILL option is given.','*23','CIRCLE','{{pt1}} {{pt2}}') newCmd('','','','','Draws a circle around pt1 in the indicated plane.','*24','CIRCLE','{{pt1}} {{plane}}') newCmd('','','','','Draws a smooth curve through the given positions. ','*25','CURVE','{{pt1}} {{pt2}} {{pt3}} ...') newCmd('','14.3.16','','*v+14.4;','Draws a curve based on the array of points.','*26','CURVE','[array of points]') newCmd('','','','','Draw CYLINDER creates a cylinder of the designated diameter. End caps can be set to closed/flat (FILL, the default) or open (MESH NOFILL).','*31','CYLINDER','{{pt1}} {{pt2}}') newCmd('','11.7.29','','*v+11.8;','Draws a frame (an x,y,z axis set) at the given center with the given quaternion orientation. Quaternions are expressed using the {#.functions~quaternion()} function within a math @{{...}} wrapper. For example, draw ID "q1" frame {{0 0 0}} @{{quaternion(1,0,1,0)}} draws a frame at the origin that has been rotated 90 degrees relative to the Y axis. (Jmol automatically normalizes the quaternion to q0=0.70710677, q1=0, q2=0.70710677, q4=0, which represents a 90-degree rotation about the Y axis.)','*34','FRAME','._atom_expression','{{quaternion}}','') newCmd('','11.7.47','','*v+11.8;','Draws a vector representing the local helical axis for the selected amino acid or nucleic acid residue, connecting it to the previous residue in its chain. The length of the arrow is the vertical height per residue. ','*35','HELIX AXIS','') newCmd('','11.9.17','','*v+12.0;','Draws the portion of a plane that intersects the current {#.boundbox~} based on a {#.plane expressions~plane expression}.','*36','INTERSECTION','BOUNDBOX','(plane expression)','') newCmd('','11.9.17','','*v+12.0;','Draws the portion of a plane that intersects the current {#.unitcell~unit cell} based on a {#.plane expressions~plane expression}.','*38','INTERSECTION','UNITCELL','(plane expression)','') newCmd('','','','','Draws a set of line segments through the points. See also DRAW VERTICES.','*39','LINE','[array of points]') newCmd('','','','','Creates a four-vertex quadrilateral even if only three points are given.','*40','PLANE','{{pt1}} {{pt2}} {{pt3}}') newCmd('','11.5.52','','*v+11.6;pg','Calculates and draws point group symmetry planes and axes for a symmetrical or nearly symmetrical molecule. As for {#.calculate~calculate pointgroup}, the calculation is carried out only on the currently selected atoms and is limited to at most 100 selected atoms. Parameters include specification of a subset to draw (Cn, C2, C3, C4, C5, C6, C8, Sn, S3, S4, S5, S6, S8, S10, S12, Cs, Ci) optionally followed by an index (for example, draw POINTGROUP C3 2 draws the second C3 axis only). A second option, SCALE x, allows adjusting the scale of the drawn planes and axes. The default scale of 1 puts the edge of planes directly through the outermost atoms. This command automatically sets perspectiveMode OFF so as to properly represent the planes and axes. ','*48','POINTGROUP','[parameters]') newCmd('','14.3.15','','*v+14.4;pg,poly','Calculates and draws point group symmetry planes and axes for a polyhedron. One atom (one polyhedron) should be selected prior to this command. ','*490','POINTGROUP','POLYHEDRON') newCmd('','14.3.15','','*v+14.4;','Draws the crystal class symmetry operations for a space group','*491','POINTGROUP','SPACEGROUP') newCmd('','11.9.19','','*v+12.0;','This POLYGON option draws one or more polygons based on a set of vertices and a set of faces. This capability allows drawing any number of flat triangular (not quadrilateral, but read on...) faces with or without edges around each face. The description is somewhat like that for PMESH files and involves (a) giving the number of vertices, (b) listing those vertices, (c) giving the number of faces, and (d) listing the faces with a special array syntax. Each face is described as an array indicating the three (0-based) vertex indices followed by a number from 0 to 7 indicating which edges to show a border on when the mesh option is given: [|| 0 | no edge|| 1| edge between first and second vertex|| 2 | edge between second and third vertex||4 | edge between third and first vertex || 3, 5, 6, 7 | combinations of the above.||] For example: draw POLYGON 4 {{0 0 0}} {{1 1 1}} {{1 2 1}} {{0 5 0}} 2 [0 1 2 6] [0 3 2 6] mesh nofill. The points and faces can be provided as arrays: draw POLYGON @points @faces mesh nofill, where in this example, points is an array of the four points, and faces is [[0 1 2 6] [0 3 2 6]].','*53','POLYGON','[polygon definition]') newCmd('','14.9.2','','*v+12.0;','This POLYGON option draws one polygon based on an array of integers (the "face") and an array of points that the indices of the face point to. The array of points is optional and defaults to {{*}}.xyz.all. For example,
:>load $p4:> x = {{*}}.find("*1**1","map"):> draw ID p4r polygon @{{x[1]}} color red:> draw ID p4b polygon @{{x[2]}} color blue:> draw ID p4y polygon @{{x[3]}} color yellow:> draw ID p4g polygon @{{x[4]}} color green
','*52','POLYGON','[array of indices]','[array of points]','') newCmd('','14.4.0','','*v+14.4;','The POINTS option creates a set of points. For example, load $caffeine; draw POINTS [@5 @7 @12 @13 @1 @3].','*50','POINTS','[array of points]') newCmd('','14.3.13','','*v+14.4;','This simple POLYGON option creates a polygon from a list of points. For example, load $caffeine; draw polygon [@5 @7 @12 @13 @1 @3].','*51','POLYGON','[array of points]') newCmd('','14.9.2','','*v+14.4;','This POLYHEDRON option creates a set of polygons, collectively forming (possibly) a polyhedron from an array of arrays of indices (that is, an array of faces) and an optional array of points, which defaults to {{*}}.xyz.all. For example, load $caffeine; draw POLYHEDRON @{{{{*}}.find("*1****1||*1*****1","map")}} fills in the 5- and 6-membered aromatic rings with a colored surface. load $p4; draw POLYHEDRON @{{{{*}}.find("*1**1","map")}} fills in the faces of tetrahedral tetraphosphorus. The faces need not be wound correctly. ','*54','POLYHEDRON','[array of arrays of atom indices]','[array of points]','') newCmd('','11.5.43','','*v+11.6;','Draws vectors for the previously selected residues representing the quaternion frame and rotational axis for each residue. The parameters for this command are the same as for {#.plot~plot quaternion} or the quaternion command. Vectors are named based on the axis (x, y, z, q), residue number, and chain. ','*6','QUATERNION','[parameters]') newCmd('','11.5.47','','*v+11.6;','Draws curved arrows marked with PHI and PSI angles in planes perpendicular to the N-CA and CA-C bonds of the selected amino acids, respectively.','*7','RAMACHANDRAN','') newCmd('','12.1.26','','*v+12.2;','Creates a DRAW object based on DRAW object $id, which must be of POLYGON type, that is slabbed based on a plane definition. For example: draw p4 polygon 4 {{0 0 0}} {{0 3 0}} {{3 3 0}} {{0 3 3}} 2 [0 1 2 0] [2 3 0 0]; draw p4 slab $p4 PLANE @{{plane({{0 0 0}}, {{5 0 0}})}}','*8','SLAB','$id','PLANE','(plane expression)') newCmd('','11.8.1','','*v+11.8;','Draws all operations in the current space group.','*901','SPACEGROUP','') newCmd('','11.8.1','','*v+11.8;show','Draws a graphic representation of a crystallographic space group symmetry operation. Operations include simple rotations, screw rotations, rotation-inversions, mirror planes, and glide planes, Either a number (for one of the model\'s symmetry operations) or a string indicating a Jones-Faithful operation may be used. The position may be an atom expression may be a point. If a point, it can be expressed either as a cartesian coordinate or a fractional coordinate (using a "/" in at least one position -- for example, {{ 1/2 0 0}}. The ID of the draw command is prepended to the drawn object IDs. For example, draw ID "s1" SYMOP "x,1/2-y,z" {{1/2 1/2 1/2}}. See also the {http://chemapps.stolaf.edu/jmol/docs/examples-11/showsym.htm~Jmol Crystal Symmetry Explorer}.','*902','SYMOP','[n or "x,y,z"]','{{atom expression}}','') newCmd('','11.9.17','','*v+12.0;show','Draws the symmetry relations between any two atoms or groups or any two positions in space. For example, draw SYMOP {{molecule=1}} {{molecule=2}}.','*91','SYMOP','{{atom expression}}','{{atom expression}}','') newCmd('','11.9.17','','*v+12.0;show','Draws the symmetry relation associated with the specified symmetry operator between any two atoms or groups or any two positions in space. For example, load =ams/quartz 1 packed;draw symop 2 @1 @3.','*921','SYMOP','(integer)','{{atom expression}}','{{atom expression}}') newCmd('','11.9.17','','*v+12.0;show','Draws the nth symmetry operator for special positions, where multiple symmetry operations relate two positions in space. For example, load =ams/quartz 1 packed;draw symop @1 @3 1.','*922','SYMOP','{{atom expression}}','{{atom expression}}','(integer)') newCmd('','11.9.3','','*v+12.0;show','Draws the symmetry operation associated with the given 4x4 matrix, which most likely comes from a variable, for example here the symmetry operation that is the product of two other symmetry operations: draw SYMOP @{{symop(11)*symop(14)}}.','*93','SYMOP','[matrix]') newCmd('','11.9.17','','*v+11.0;','Draws the currently defined {#.unitcell~}. Note that by default this is a solid. To show just edges, use options MESH NOFILL.','*94','UNITCELL','') newCmd('','','','','This option is similar to ARROW, and accepts two points. The first point is the origin; the second is of the form {{dx,dy,dz}}, indicating a vector to the next point rather than the point itself.','*95','VECTOR','{{pt1}} {{pt2}}') newCmd('.echo','11.1.6','','label;math','In Jmol, models can be annotated in one of three ways. Text can be associated with a specific atom using a {#.label~}, text can appear when the user hovers the mouse over an atom or other user-defined point in space for a designated period of time ({#.hover~}), and text can be placed at a specified position on the window (2D echo) or at a point in space (3D echo). In addition, the text is echoed in the Java console, the Jmol {#.console~}, and the {#.set_messageCallback~} or {#.set_echoCallback~} function, if defined. Multi-line text can be generated using a vertical bar as a line separator.
See also the {#.message~} command for variable-displaying capabilities that send information to the consoles and callback functions without displaying text. One can also place JPEG, PNG, or GIF images at either a 2D screen position or a 3D molecular position. See {#.set echo~} for details. ','0|1','','') newCmd('','11.1.6','','label;math','Sends text to the console, callbacks, and, depending upon the setting of {#.setecho~set echo}, to a position in the Jmol window. Echos can contain variables such as @x. If in an echo is one that is displayed and the string contains a variable expression in quotes, such as set echo top center; echo "@{{_modelNumber}}", the echo is updated dynamically every time the screen is refreshed. Variable substitution can be prevented using \\@ instead of @.','1','(string)','') newCmd('.else','','','*v+11.4;flow','See {#.if~}.','0|1','','') newCmd('.elseIf','','','*v+11.4;flow','See {#.if~}.','0|1','','') newCmd('.ellipsoid','11.5.29','','*v+11.6 NEW;disp','The ellipsoid command displays anisotropic displacement parameters (thermal ellipsoids) based on crystallographic Uij data in CIF or PDB files and tensor quantities found in CRYSTAL files. (For PDB files, you must specify a lattice, for example, load =1a6g {{1 1 1}}, in order to have Jmol read the ANISOU records.) The resulting ellipsoids have three mutually perpendicular axes that are not necessarily the same length. Overall ellipsoid size can be set to a percentage value (default 50%). Rendering styles are set globally using the set command and one or more of the settings described below. The diameter of the axis and arc lines can be set using set ellipsoidAxisDiameter x.x where x.x is a distance in Angstroms (default 0.02). Note that the Uij data when read from CIF files are also stored in the .temperature value for atoms as 100 times the geometric mean of the diagonal U-factor parameters, 100 * (U11 * U22 * U33)^0.3333.
User-defined ellipsoids can also be created by specifying the keyword ID followed by a user-named ID. These parameters may be indicated in any order, but the ellipsoid is not defined until the AXES parameter is specified.
[||set ellipsoidAxes|
 This option may be combined with any other option. When combined with balls, the color of the axes are set to white or black in order to contrast with the background.||set ellipsoidArcs|
This option may be combined with any other option. When combined with balls, the color of the axes are set to white or black in order to contrast with the background.||set ellipsoidArcs| When combined with balls, the color of the arcs are set to white or black in order to contrast with the background. This option is ignored when dots are displayed.||set ellipsoidBall|
When combined with balls, the color of the arcs are set to white or black in order to contrast with the background. This option is ignored when dots are displayed.||set ellipsoidBall| This option is the default option and may be combined with ellipsoidAxes, ellipsoidArcs, or ellipsoidFill (which displays a cut-out octant).||set ellipsoidFill|
This option is the default option and may be combined with ellipsoidAxes, ellipsoidArcs, or ellipsoidFill (which displays a cut-out octant).||set ellipsoidFill|

 This option affects the rendering of arcs and balls. In the case of arcs, it fills in the arcs to form a set of three elliptical planes; in the case of balls, it provides a cut-out octant. When combined with {#.spacefill~spacefill ADPMIN n%}, where n is the current percentage size of the ellipsoid, the combination of fill and arcs provides a strikingly unique rendering of the anisotropic displacement parameters for an atom. ||set ellipsoidDots|
This option affects the rendering of arcs and balls. In the case of arcs, it fills in the arcs to form a set of three elliptical planes; in the case of balls, it provides a cut-out octant. When combined with {#.spacefill~spacefill ADPMIN n%}, where n is the current percentage size of the ellipsoid, the combination of fill and arcs provides a strikingly unique rendering of the anisotropic displacement parameters for an atom. ||set ellipsoidDots| This option renders a random set of dots defining the ellipsoid that sparkle as the model is manipulated. The number of dots can be set using set ellipsoidDotCount n where n is an integer (default 200). This option is ignored when ellipsoid balls are displayed.||]','0|1','','')
newCmd('','','','*v+11.6;','Turns ellipsoids on or off for the currently selected atoms.','*1','._on_off{"ON"}','')
newCmd('','','','*v+11.6;','Sets the size of the ellipsoids for the currently selected atoms.','*2','nn%','')
newCmd('','','','*v+11.6;','Sets the three perpendicular axes for the ellipsoid. These axes should be perpendicular. If they are not, the ellipsoid may not be rendered, or its shape will be unpredictable. The ellipsoid is not displayed until this parameter is set.','*35','ID','[object id]','AXES','{{ax ay az}} {{bx by bz}} {{cx cy cz}}')
newCmd('','','','*v+11.6;','Sets the color of the ellipsoid using parameters described in the {#.color~} command. ','*5','ID','[object id]','COLOR','[color parameters]')
newCmd('','','','*v+11.6;','Sets the scale of the ellipsoid relative to its axes lengths.','*8','ID','[object id]','SCALE','(decimal)')
newCmd('','','','*v+11.6;','Deletes the specified ellipsoid.','*6','ID','[object id]','DELETE','')
newCmd('','13.1.19','','*v+13.2;','Sets one or more options separated by semicolon: "arcs;arrows;axes;ball;dots;fill;wireframe" with optional "no" in front of one or more. This option overrides the settings of set_ellipsoidball and related settings.','*7','ID','[object id]','OPTIONS','"options"')
newCmd('','','','*v+11.6;','Sets the center for this ellipsoid the specified point (which may be fractional).','*41','ID','[object id]','CENTER','{{x y z}}')
newCmd('','','','*v+11.6;','Sets the center for this ellipsoid to the center of the specified atoms.','*42','ID','[object id]','CENTER','{{ atom expression }}')
newCmd('','','','*v+11.6;','Sets the center for this ellipsoid to the point specified by the object name (draw or ellipsoid)','*43','ID','[object id]','CENTER','$object')
newCmd('','','','*v+11.6;','Turns this ellipsoid on.','*31','ID','[object id]','ON','')
newCmd('','','','*v+11.6;','Turns this ellipsoid off.','*32','ID','[object id]','OFF','')
newCmd('.[export]','','','*v+11.4;','The Jmol application (not the applet) allows export of the currently rendered scene as files that can be read by {http://www.avatech.com/solutions/visualization/product-details.aspx?product=17~Maya}, {http://www.povray.org/~POV-Ray}, and VRML readers. See the {#.write~} command for details.','0','','')
newCmd('.exit','','','*v+11.0 -- new;pause;quit','When the {#.set_scriptQueue~} is turned on, each script waits for the previous to complete. Either {#.quit~} or exit at the very beginning of a script command halts any previous still-running script. Processing then continues with the second command on the line. Anywhere else in the command, quit and exit abort that script only. In addition, exit clears the script queue of any remaining scripts, thus stopping all script processing.','0','','')
newCmd('.file','','','anim','Carries out a {#.frame~} command bringing into frame all models in the specified file number.','1','(integer)','')
newCmd('.fix','','','','The fix command takes argument like display or select. But ensures no atoms of this set will be moved or dragged anywhere accidentally.','1','._atom_expression','')
newCmd('.font','','','label','Sets font size, face (serif, sansSerif), and style (plain, italic, bold, bolditalic) in {#.label~labels} and other text-bearing elements, including {#.axes~}, {#.echo~}, {#.hover~}, and {#.measure~}. If only a font size is given and the object is a label, then only the size of the font is changed, not the font face or style. For all other objects, if only a font size is given, the font face and style are changed to sansSerif. For font ECHO you can apply a scaling factor that allows the font to scale with zoom. This scaling factor, in "pixels per micron", determines the absolute size of the font relative to a "standard" window size. (Scaled {#.echo~} fonts can also be created using set fontscaling TRUE, then defining the echo text. The scaling factor also applies to images added with the {#.set echo IMAGE~set echo} command. Once a {#.label~} font size is set with a font scaling factor, if it is necessary to change the label and not its size and the zoom is not 100, it is necessary to use {#.set fontscaling~} OFF just before changing the label and then back ON just after. This is because {#.set fontscaling~} always uses the current zoom as part of its calculation.','5','._object_with_text','._fontsize','._fontface{"SansSerif"}','._fontstyle{"Plain"} [scaling factor]')
newCmd('.for','','','*v+12.2 -- for (var x IN ...);math;flow','Jmol supports both the standard for statement as well as a specialized in-line FOR construct that provides a powerful way to work with atom data.','0|1','','')
newCmd('.for','','','*v+11.4 -- new flow control options;*v-12.2;math;flow','Jmol supports both the standard for statement as well as a specialized in-line FOR construct that provides a powerful way to work with atom data.','0|1','','')
newCmd('','','','*v+11.4;math;flow','for takes three aguments in parentheses and separated by a semicolon. The loop may be exited using {#.break~} and truncated using {#.break~continue}. For example, for(var i=1; i <= 10; i++){{...}} loops through a block of script ten times, incrementing the variable i by one each time. Any variables created with the VAR keyword are local to the for block.','TEXT','for (var i = 1; i < n; i++) {...}','')
newCmd('','14.3.13','','*v+14.4;math;flow','A second form of for takes two arguments -- a variable name and a two-element array defining the integer range to loop over. For example, for (var i FROM [1,n]) {{. . .}}. This allows for faster loop processing. Variable b can be larger or smaller than a, allowing the loop variable to increase or decrease each loop, respectively.','TEXT','for (var i FROM [a,b]) {...}','')
newCmd('','','','*v+12.2 -- new;math;flow','A third form of for takes just two arguments -- a variable name and either an atom set or an array. For example, for(var i IN {{selected}}) {{print i.label()}} loops through all atoms that have been selected, assigning i to each one in turn, printing its default label, and var a = [100,150,200]; for(var i IN a) {{print i + 5}} prints a list that is five more than the array values. The loop may be exited using {#.break~} and truncated using {#.break~continue}. If the looping variable (i in this case) is assigned any value during the process, the for loop will exit at the end of the current loop. Thus, for example, i = 0 is a way of saying: "Continue to the end of this loop and then exit."','TEXT','for (var i IN [atom-expression or array]) {...}','')
newCmd('','11.7.40','','*v+11.8;','You can create lists of values using a form of inline FOR function. The syntax is simply xxx = for(dummyVariable;{{atom expression}};math expression). For example,
This option renders a random set of dots defining the ellipsoid that sparkle as the model is manipulated. The number of dots can be set using set ellipsoidDotCount n where n is an integer (default 200). This option is ignored when ellipsoid balls are displayed.||]','0|1','','')
newCmd('','','','*v+11.6;','Turns ellipsoids on or off for the currently selected atoms.','*1','._on_off{"ON"}','')
newCmd('','','','*v+11.6;','Sets the size of the ellipsoids for the currently selected atoms.','*2','nn%','')
newCmd('','','','*v+11.6;','Sets the three perpendicular axes for the ellipsoid. These axes should be perpendicular. If they are not, the ellipsoid may not be rendered, or its shape will be unpredictable. The ellipsoid is not displayed until this parameter is set.','*35','ID','[object id]','AXES','{{ax ay az}} {{bx by bz}} {{cx cy cz}}')
newCmd('','','','*v+11.6;','Sets the color of the ellipsoid using parameters described in the {#.color~} command. ','*5','ID','[object id]','COLOR','[color parameters]')
newCmd('','','','*v+11.6;','Sets the scale of the ellipsoid relative to its axes lengths.','*8','ID','[object id]','SCALE','(decimal)')
newCmd('','','','*v+11.6;','Deletes the specified ellipsoid.','*6','ID','[object id]','DELETE','')
newCmd('','13.1.19','','*v+13.2;','Sets one or more options separated by semicolon: "arcs;arrows;axes;ball;dots;fill;wireframe" with optional "no" in front of one or more. This option overrides the settings of set_ellipsoidball and related settings.','*7','ID','[object id]','OPTIONS','"options"')
newCmd('','','','*v+11.6;','Sets the center for this ellipsoid the specified point (which may be fractional).','*41','ID','[object id]','CENTER','{{x y z}}')
newCmd('','','','*v+11.6;','Sets the center for this ellipsoid to the center of the specified atoms.','*42','ID','[object id]','CENTER','{{ atom expression }}')
newCmd('','','','*v+11.6;','Sets the center for this ellipsoid to the point specified by the object name (draw or ellipsoid)','*43','ID','[object id]','CENTER','$object')
newCmd('','','','*v+11.6;','Turns this ellipsoid on.','*31','ID','[object id]','ON','')
newCmd('','','','*v+11.6;','Turns this ellipsoid off.','*32','ID','[object id]','OFF','')
newCmd('.[export]','','','*v+11.4;','The Jmol application (not the applet) allows export of the currently rendered scene as files that can be read by {http://www.avatech.com/solutions/visualization/product-details.aspx?product=17~Maya}, {http://www.povray.org/~POV-Ray}, and VRML readers. See the {#.write~} command for details.','0','','')
newCmd('.exit','','','*v+11.0 -- new;pause;quit','When the {#.set_scriptQueue~} is turned on, each script waits for the previous to complete. Either {#.quit~} or exit at the very beginning of a script command halts any previous still-running script. Processing then continues with the second command on the line. Anywhere else in the command, quit and exit abort that script only. In addition, exit clears the script queue of any remaining scripts, thus stopping all script processing.','0','','')
newCmd('.file','','','anim','Carries out a {#.frame~} command bringing into frame all models in the specified file number.','1','(integer)','')
newCmd('.fix','','','','The fix command takes argument like display or select. But ensures no atoms of this set will be moved or dragged anywhere accidentally.','1','._atom_expression','')
newCmd('.font','','','label','Sets font size, face (serif, sansSerif), and style (plain, italic, bold, bolditalic) in {#.label~labels} and other text-bearing elements, including {#.axes~}, {#.echo~}, {#.hover~}, and {#.measure~}. If only a font size is given and the object is a label, then only the size of the font is changed, not the font face or style. For all other objects, if only a font size is given, the font face and style are changed to sansSerif. For font ECHO you can apply a scaling factor that allows the font to scale with zoom. This scaling factor, in "pixels per micron", determines the absolute size of the font relative to a "standard" window size. (Scaled {#.echo~} fonts can also be created using set fontscaling TRUE, then defining the echo text. The scaling factor also applies to images added with the {#.set echo IMAGE~set echo} command. Once a {#.label~} font size is set with a font scaling factor, if it is necessary to change the label and not its size and the zoom is not 100, it is necessary to use {#.set fontscaling~} OFF just before changing the label and then back ON just after. This is because {#.set fontscaling~} always uses the current zoom as part of its calculation.','5','._object_with_text','._fontsize','._fontface{"SansSerif"}','._fontstyle{"Plain"} [scaling factor]')
newCmd('.for','','','*v+12.2 -- for (var x IN ...);math;flow','Jmol supports both the standard for statement as well as a specialized in-line FOR construct that provides a powerful way to work with atom data.','0|1','','')
newCmd('.for','','','*v+11.4 -- new flow control options;*v-12.2;math;flow','Jmol supports both the standard for statement as well as a specialized in-line FOR construct that provides a powerful way to work with atom data.','0|1','','')
newCmd('','','','*v+11.4;math;flow','for takes three aguments in parentheses and separated by a semicolon. The loop may be exited using {#.break~} and truncated using {#.break~continue}. For example, for(var i=1; i <= 10; i++){{...}} loops through a block of script ten times, incrementing the variable i by one each time. Any variables created with the VAR keyword are local to the for block.','TEXT','for (var i = 1; i < n; i++) {...}','')
newCmd('','14.3.13','','*v+14.4;math;flow','A second form of for takes two arguments -- a variable name and a two-element array defining the integer range to loop over. For example, for (var i FROM [1,n]) {{. . .}}. This allows for faster loop processing. Variable b can be larger or smaller than a, allowing the loop variable to increase or decrease each loop, respectively.','TEXT','for (var i FROM [a,b]) {...}','')
newCmd('','','','*v+12.2 -- new;math;flow','A third form of for takes just two arguments -- a variable name and either an atom set or an array. For example, for(var i IN {{selected}}) {{print i.label()}} loops through all atoms that have been selected, assigning i to each one in turn, printing its default label, and var a = [100,150,200]; for(var i IN a) {{print i + 5}} prints a list that is five more than the array values. The loop may be exited using {#.break~} and truncated using {#.break~continue}. If the looping variable (i in this case) is assigned any value during the process, the for loop will exit at the end of the current loop. Thus, for example, i = 0 is a way of saying: "Continue to the end of this loop and then exit."','TEXT','for (var i IN [atom-expression or array]) {...}','')
newCmd('','11.7.40','','*v+11.8;','You can create lists of values using a form of inline FOR function. The syntax is simply xxx = for(dummyVariable;{{atom expression}};math expression). For example, :>>aniso = for(x;{{*}};x.adpmax - x.adpmin)creates an array containing differences between the maximum and minimum anisotropic density parameters for a set of atoms. Arrays such as these can be used to assign properties to atoms or to color them. So, for example, we might want to set the radius of atoms based on temperature, but not the exact value of the temperature:
:>>{{*}}.radius = for(x;{{*}};x.temperature/20)
Combined with the inline IF function, the inline FOR can be quite useful. For example:
:> {{*}}.color = for(x;{{*}};if(x.temperature > 10;"red";if(x.temperature < 2;"blue";"white")))
See also {#.functions~} for how to define a function that operates on atoms one at a time with the syntax
x = getproperty("modelinfo.models.energy")
y = x.sub(x.min).mul(2625.5) # relative energies, in kJ
frame *;frame title @y','*93','TITLE','(array)') newCmd('.frank','','frank','*v+11.0 -- new;','Determines whether or not "Jmol" is indicated in the bottom right corner of the window.','0|1','','') newCmd('','','','*v+11.0;','Turning the frank off is disabled for the signed applet running on a web server.','1','._on_off','') newCmd('.geoSurface','','','*v+11.0 -- provides a quickly rendered crude geodesic molecular/solvent-accessible surface.;disp','Turns a crude geodesic molecular surface on or off around the currently selected atoms. If a decimal with an explicit "+" sign is given, or {#.set_solvent~set solvent ON}) is in effect, the resultant surface is a crude solvent-accessible surface. This command has the same syntax as the {#.dots~} command. To color the surface, use {#.color (model object)~color geosurface}. For a smoother surface, use {#.isosurface~isosurface SASURFACE 1.2}.','*11','._on_off{"ON"}','') newCmd('','','','*v+11.6;','Turns geosurface rendering on and all other rendering (including labels) off.','*12','ONLY','') newCmd('','','','*v+11.0;','Draws a geodesic surface at the van der Waals radius for the selected atoms. See {misc/radii.xls~radii.xls}.','*2','VANDERWAALS','') newCmd('','','','*v+11.0;','Draws a geodesic surface at the (nominal) ionic radius for the selected atoms if the atom\'s charge has been read and is nonzero; uses the nominal covalent bonding radius otherwise ({misc/radii.xls~radii.xls}).','*3','IONIC','') newCmd('','','','*v+11.0;','Draws a geodesic surface at the indicated percent of the van der Waals radius for each atom (maximum value 1000%).','*4','(integer)','') newCmd('','','','*v+11.0;','Draws a geodesic surface at the indicated radius in Angstroms for each atom (maximum value 10.0 Angstroms).','*5','(decimal)','') newCmd('','','','*v+11.0;','Draws a geodesic surface at the indicated distance in Angstroms beyond the van der Waals radius for each atom (maximum value 10.0 Angstroms). The "+" sign is required. This surface approximates the solvent-accessible surface with the indicated solvent probe radius. Typically this number is +1.2 or +1.4. This command overrides the {#.set_solvent~set solventProbe/set radius} method of defining the solvent-accessible surface.','*6','+(decimal)','') newCmd('.getProperty','','','*v+11.0 NEW;javascript','The getProperty command sends information to the {#.console~Jmol console} or message callback function defined for a Jmol applet using the jmolSetCallback("messageCallback", funcName) function in Jmol.js or via the {#.set (callback)~set} command. Either a simple text string in the case of a file property or a valid JSON (JavaScript Object Notation) string in the case of a molecular property is returned. Such callbacks are asynchronous, meaning they do not return a value immediately. An alternative synchronous method for the applet is to use one of the JSmol built-in JavaScript commands Jmol.getPropertyAsArray(), Jmol.getPropertyAsString(), Jmol.getPropertyAsJSON(), or Jmol.getPropertyAsJavaObject(), all of which take three parameters: applet, infoType, and params. For example, in JavaScript:
var modelInfo = Jmol.getPropertyAsArray(jmolApplet0, "modelInfo")
alert(modelInfo.modelCount)
for (int i = 0; i < modelInfo.modelCount; i++):>alert(modelInfo.models[i].name)In addition, when using the Jmol application, see the note at {#.show~} regarding setting the output to go directly into a file on your system.
This information is also available using the Jmol math {#.functions~getProperty()} function.','0|1','','') newCmd('','','','*v+11.0;','Structure describing the current state of animation. See {misc/animationInfo.txt~animationInfo.txt}.','*10','animationInfo','') newCmd('','','','*v+11.0;','Structure describing the applet, including, for example, the applet version, compile date, Java version, and name of the applet object. See {misc/appletInfo.txt~appletInfo.txt}.','*11','appletInfo','') newCmd('','','','*v+11.0;','Structure describing the atoms in the model. A second (optional) parameter specifies a subset of the atoms. The default is (visible). Parentheses are required. See {misc/atomInfo.txt~atomInfo.txt}.','*12','atomInfo','._atom_expression') newCmd('','','','*v+11.0;','Structure describing auxiliary information that is in the loaded file. This information is file-dependent and might include, for example, symmetry information, molecular orbital coefficients, dipole moments, partial charges, and/or vibrational modes. See {misc/auxiliaryInfo.txt~auxiliaryInfo.txt}.','*14','auxiliaryInfo','') newCmd('','','','*v+11.0;','Structure describing the bonds in the model. A second (optional) parameter specifies a subset of the atoms that are involved in the bonds. The default is (visible). Parentheses are required. See {misc/bondInfo.txt~bondInfo.txt}.','*15','bondInfo','._atom_expression') newCmd('','','','*v+11.0;boundbox','A simple JSON array containing the coordinates of the center and corner of the volume containing the molecule. See {misc/boundBoxInfo.txt~bondBoxInfo.txt}.','*16','boundBoxInfo','') newCmd('','','','*v+11.0;','A single JSON array giving the current center coordinate. See {misc/centerInfo.txt~centerInfo.txt}.','*17','centerInfo','') newCmd('','','','*v+11.0;','Structure describing the chains in a biomodel (PDB or mmCIF, for example). Information for each residue of the chain is provided. A second (optional) parameter specifies a subset of the atoms. The default is (visible). Parentheses are required. See {misc/chainInfo.txt~chainInfo.txt}.','*180','chainInfo','._atom_expression') newCmd('','','','*v+11.0;','The contents of a CIF file in structured data format or for the current file if no filepath is given.','*185','cifInfo','filepath') newCmd('','','','*v+11.0;','Returns an array having two elements. Generally the first element is the data label, and the second element is the {#.data~} itself. getProperty data TYPES returns a first element that is the string "types" and a second element that is a comma-separated list of the available data types. ','*19','dataInfo','type') newCmd('','','','*v+11.0;','The extractModel keyword delivers text in the form of a MOL file, allowing up to 999 atoms and 999 bonds to be "extracted" from the model as an independent structure. ','*20','extractModel','._atom_expression') newCmd('','','','*v+11.0;','The contents of the currently loaded file.','*21','fileContents','') newCmd('','','','*v+11.0;','The contents of ANY file on the web or, if operating locally, any file on the hard drive in the directory containing the JAR file or any directory below that.','*22','fileContents','filepath') newCmd('','','','*v+11.0;','The file header for the file. This will depend upon the file format; some file formats may not have file headers.','*23','fileHeader','') newCmd('','','','*v+11.0;','The file name of the currently loaded file.','*241','fileName','') newCmd('','','','*v+11.0;','An associative array of file information. For most files this is simply the file header lines. In the case of PDB files, this array associates PDB header information with its resource name. For example, getProperty fileInfo.SHEET reports SHEET information from the PDB header. In the case of CIF files, this information is the entire contents of the file in array format. (See also the function getProperty("fileInfo"), which allows direct access to file information as a Jmol variable. For example, x = getProperty("fileInfo.REMARK800"). ','*243','fileInfo','type') newCmd('','','','*v+11.0;','A base-64 encoded JPEG suitable for writing by the applet to an image tag using a dataURI. For example, in JavaScript on the page using JSmol.min.js, with an applet named jmolApplet0: var img = new Image(); img.src="data:image/jpeg;base64," + Jmol.getPropertyAsString(jmolApplet0,"image");$("body").append(img) .','*251','image','') newCmd('','13.1.7','','*v+13.2;','Returns information about an isosurface of getProperty isosurfaceInfo along with vertices, vertex values, and polygons.','*252','isosurfaceData','') newCmd('','','','*v+12.2;','Returns information about an isosurface, including haveQuads, haveValues, id, polygonCount, and vertexCount.','*253','isosurfaceInfo','') newCmd('','','','*v+12.2;','Structure indicating ligand information, including and array of "ligands", each containing items "reslist", "atoms", and "groupNames".','*281','ligandInfo','') newCmd('','','','*v+11.0;','Structure describing the currently-defined measurements for the model, including the atoms involved, the measurement type, and the value of the measurement in decimal and in string formats. See {misc/measurementInfo.txt~measurementInfo.txt}.','*282','measurementInfo','') newCmd('','','','*v+11.4;','A tab-separated string defining the current Jmol menu, including what options are currently enabled. See, for example, {misc/menu.tab~misc/menu.tab}. See also {#.show~show MENU}.','*300','menu','') newCmd('','11.5.23','','*v+11.6;','A summary of the minimization that was carried out. See {misc/minimizationInfo.txt~minimizationInfo.txt}.','*301','minimizationInfo','') newCmd('','','','*v+11.0;','Structure describing each model in the loaded model collection. See {misc/modelInfo.txt~modelInfo.txt}.','*302','modelInfo','') newCmd('','','','*v+11.0;','Structure describing each molecule (covalent set of atoms) in the model, including the number of atoms, the number of elements, and the molecular formula. A second (optional) parameter specifies a subset of the atoms. The default is (visible). Parentheses are required. See {misc/moleculeInfo.txt~moleculeInfo.txt}.','*311','moleculeInfo','._atom_expression') newCmd('','13.1.19','','*v+13.2;','Structure describing NMR calculation data including (a) a table of isotope masses (negative if these are default parameters for a nucleus), gyromagnetic ratio, and quadrapolar coupling constant, and (b) assigned shift references in PPM. See {misc/nmrInfo.txt~nmrInfo.txt}.','*312','nmrinfo','') newCmd('','','','*v+11.0;','Structure describing the moveTo command required to return to the currently displayed orientation. See {misc/orientationInfo.txt~orientationInfo.txt}.','*32','orientationInfo','') newCmd('','','','*v+11.0;','Structure similar to chainInfo describing the residues in a biomodel. A second (optional) parameter specifies a subset of the atoms. The default is (visible). Parentheses are required. See {misc/polymerInfo.txt~polymerInfo.txt}.','*33','polymerInfo','._atom_expression') newCmd('','','','*v+11.0;','Structure listing a small amount of information relating to shapes displayed with the model (molecular orbitals, isosurfaces, cartoons, rockents, dipoles, etc.) See {misc/shapeInfo.txt~shapeInfo.txt}.','*34','shapeInfo','') newCmd('','','','*v+11.0;','The same report as SHOW STATE; useful especiall as x = getProperty("stateInfo").','*35','stateInfo','') newCmd('','','','*v+11.0;','Structure representing the current transformation matrix describinng the current orientation of the model. See {misc/transformInfo.txt~transformInfo.txt}.','*36','transformInfo','') newCmd('','','','*v+14.2;','Evaluates a variable. This option is most likely used in association with JavaScript. The JSmol API includes Jmol.evaluateVar(app,expr) where app is an applet object and expr is a string containing a Jmol math expression (anything that work with the print command). For example, "{atomno < 3}" or "at.join(af)".','*37','variableInfo','(math expression)') newCmd('.goto','11.1.9','','*v+11.2 -- new;pause;flow','Transfers script execution to the {#.message~} command having the matching text. If an underscore is at the beginning of the text, then the text is not displayed, otherwise the message text is displayed in the console as usual for that command.
message _test
...
...
goto _test
The goto command is largely deprecated by {#.if_while_for~if, while, and for}. These alternative commands are recommended.','0|1','','') newCmd('.halos','','','*v+11.6 -- adds new capability;halos','Displays a translucent two-dimensional ring around an atom. Halos are similar to {#.star~stars}, except halos may also be used for automatically displaying which atoms are currently selected. (This option is enabled using {#.selectionHalos~}.) The radius can be set using the same options as for {#.spacefill~}, but the actual radius of the halo will always be from 4 to 10 pixels larger then the nominal radius. ','0|1','','') newCmd('','','','','Turn halos on or off. The radius will change with the current spacefill setting.','*1','._on_off{"ON"}','') newCmd('','','','*v+11.6;','Turns halo rendering on and all other rendering (including labels) off.','*12','ONLY','') newCmd('','','','','Resets the radius of the halo to the default {#.spacefill~} radius.','*4','reset','') newCmd('.help','','','*v+11.0 -- new;','Opens a new browser window to the interactive help page. See also {#.set(misc)~set helpPath}. Currently for the applet only; the query will be searched as an exact, complete phrase (applet only).','1','query','') newCmd('.hbonds','','','*v+12.0 -- full support for hydrogen bonds;bond;hbondc;hbond;calc','Hydrogen bonds can be turned on or off, given custom widths in Angstroms, or colored (see {#.color(bond object)~color hbonds} and {#.set(bond styles)~set HBONDS} ). In addition, Jmol allows calculation of actual hydrogen bonds (when hydrogen atoms are present in the selected set) or "pseudo" hydrogen bonds if hydrogen atoms are not present.','0|1','','') newCmd('','','','','','*1','._on_off{"ON"}','') newCmd('','','','','','*2','._hbond_width_angstroms','') newCmd('','','','*v+12.0;calc','Calculates hydrogen bonds involving atoms currently selected and displays them. See {#.calculate~calculate HBONDS} for details.','*4','CALCULATE','') newCmd('','','','*v+12.0;calc','Deletes hydrogen bonds between atoms in the currently selected set of atoms. Both atoms of a given hydrogen bond need to be selected. ','*5','DELETE','') newCmd('.hide','','','*v+12.0 -- hide/display bonds;dhide','The opposite of {#.display~}. Hides atoms and associated structures (bonds, halos, stars, cartoons, etc.). Hide is similar to {#.select~} or {#.restrict~} in its syntax. Unlike restrict, though, hide is completely reversible using hide none. (Restrict acts by setting the "size" of the object to 0; hide leaves the size the same, but just hides the object until unhidden. Group-based structures such as {#.cartoon~cartoons} and traces are hidden whenever their lead atom (the one that determines their position in space) is hidden. Hidden atoms can be selected with select hidden or deselected with select not hidden. Atoms can be added to the hidden set using hide hidden or ... or removed from the hidden set using hide hidden and not ..... In addition, bonds can be hidden or displayed.','0|1','','') newCmd('','','','','Hides a set of atoms. Displays atoms and associated structures (bonds, halos, stars, cartoons, etc.) and hides all others. The optional parameters ADD and REMOVE prior to the atom expression allow quick alternatives to "hidden or..." and "hidden and not..." In addition, the keyword GROUP prior to the atom expression provides a quick alternative to "within(group, ...)".','1','._atom_expression','') newCmd('','','','','Hides a set of bonds.','2','[{{...}}]','') newCmd('','','','','Hides all bonds.','3','BONDS','') newCmd('.history','','','*v+11.0 -- new;','The history command turns command history recording on and off. Each time it is invoked, the command history list is cleared. See also {#.set(files and scripts)~set history} and {#.show~show history}.','1','._on_off{"ON"}','') newCmd('.hover','','','*v+11.0 -- adds hoverCallback and multiline hover;label','Turns on and off pop-up labels that appear when the user "hovers" the mouse over the atom or a point associated with a {#.draw~} object. hover off disables the hover message; hover on enables it again. Any other parameter or string is used as the label, which can contain atom property fields such as %a or %U and will apply to all atoms in the model. To set a specific hover label for one atom, select the atom(s) and then use {#.set_hoverLabel~set hoverLabel}. To change the delay time prior to the label appearing, use set hoverDelay x.x where x.x is the delay time in seconds. Setting this time to 0.001 seconds effectively makes the hover label appear immediately. See also {#.label~}. Multiple lines can be indicated using | (vertical bar). Even with hover OFF, the hover message can be sent to a JavaScript function on the applet\'s page using {#.set_hoverCallback~}.','1|2','._on_off_string','') newCmd('.if','11.7.40','','*v+11.4;math;flow','Jmol scripting allows for both the standard if/else/elseif statement set as well as the standard Java/JavaScript-like inline (ifThis ? thenThis : elseThis) construct.','1|2','','') newCmd('','11.7.40','','*v+11.4;math;flow','The if statement takes a logical expression of variables and evaluates it either as TRUE or FALSE. Tests include a wide variety of flags that can be set using the {#.set~set} command as well as any {#.Jmolmath~user-defined variables}. The standard commands elseif (or else if) and else are supported. If blocks are defined by matched pairs of braces:
if (doSpacefill) {{:>spacefill only
}} elseif (doWireframe) {{:>wireframe only
}} else {{:>wireframe 0.15;spacefill 20%
}}
These constructs are compatible with {#.goto~}. However, using goto to jump into the middle of an if, while, or for block is considered bad form and is not recommended.
Note that with the Jmol application multiline scripts are not allowed on the script window command line. Either separate commands by semicolon and introduce them all on the same line, or store the script in a file and run it using the {#.script~} command. This is not an issue with the applet.
if/else/endif syntax is also supported. If/else/endif syntax can be entered on a single line without unnecessary semicolons:
:>>if (this) print "this" else print "that":>>if (this) {{ print "this" }} else {{ print "that" }} :>>if (this); print "this"; else; print "that"; endif;
are all identical.','TEXT','if/else/elseif','') newCmd('','11.7.42','','*v+11.8;','You can use a similar construct to Java\'s/JavaScript\'s (ifTrue ? thisValue : orThisTrue ? thatValue : otherValue). Jmol accepts this format provided the operator "?" is separated by spaces. Jmol also allows for a syntax that uses semicolons:
:>>xxx = if(ifTrue; thisValue; thatValue)
For example,
:>>set echo top left;echo @{{(_modelnumber > 1.1 ? "previous" : "next")}}
or, equivalently,
:>>set echo top left;echo @{{if(_modelnumber > 1.1;"previous" ; "next")}}
. Used in conjunction with inline FOR, the inline IF provides a powerful and concise way to work with molecular data.','TEXT','in-line IF','') newCmd('.image','14.3.13','','*v+14.4;','The IMAGE command can be used to display arbitrary 2D images. In Java, these images are brought up in their own windows; in HTML5, you have the option to open a new floating div or to designate page-based divs in which to hold specific images. In HTML5, the image will be placed in (a) a div with id (appletID)_Image_(id)_holder or, if that does not exist, (b) a div with id (appletID)_Image_holder, or if that does not exist, (c) a floating image div with ID (appletID)_Image_(id) that contains the div (appletID)_Image_(id)_holder. (appletID) is the applet id, for example, jmolApplet0; (id) is from the specified ID, or the file name if that is given, or "app" if it is not. This ID is "cleaned" by removing the path of a file name and replacing all nonalphanumeric characters with "_". ','0|1','','') newCmd('','14.3.13','','','Displays the current image. Specifying an ID is optional, defaulting to "app". ','*1','ID','id') newCmd('','14.3.13','','','Allows display of the current image at a different width or height','*2','ID','id','width','height') newCmd('','14.3.13','','','Displays a file-based image.','*3','ID','id','"filename"','') newCmd('','14.3.13','','','Closes the specified image window.','*4','ID','id','close','') newCmd('','14.3.13','','','Closes the image window associated with the given file, assuming an ID has not been specified.','*5','"filename"','close') newCmd('','14.3.13','','','Closes all image windows.','*6','close','') newCmd('.initialize','','','*v+11.2 -- new;load;rreset','The initialize command is a specialized script command only intended for use within a state script created using the {#.write~write state} or {#.save~save state} commands. It deletes all user variables, including those created using the {#.define~DEFINE} command, and resets most settings to original Jmol values in an anticipation of those settings being set in the state. It does not do a full reset to Jmol\'s initial state, however. If that capability is desired, one should save the initial state and then {#.restore~} it. Settings specifically NOT set include: allowGestures, allowKeyStrokes, allowModelkit, allowMultiTouch, debugScript, defaultDirectory, disablePopupMenu, legacyAutoBonding, messageStyleChime, useScriptQueue.','0|1','','') newCmd('.invertSelected','11.1.13','','*v+11.2 -- new;anim','This command allows inversion of atoms through a point, across a plane, or at a chirality center. With no parameters, the model is inverted through its geometric center.','0|1','','') newCmd('','11.1.13','','*v+11.2;','Following the POINT keyword may be any expression that can be evaluated to give a point, including {{x y z}} (cartesian or fractional), a draw object identifier, $xxx, or an atom expression in parentheses, which will be evaluated as an average position. Only the currently selected atoms will be inverted.','*4','POINT','point_definition') newCmd('','11.1.13','','*v+11.2;','Inverts the currently selected atoms across a plane defined using any valid {#.plane expressions~plane expression}. ','*3','PLANE','plane_expression') newCmd('','11.1.13','','*v+11.2;','Inverts the currently selected atoms across a plane, using Miller indices to specify the plane.','*2','HKL','{{h k l}}') newCmd('','14.5.2','','*v+14.5;','Carries out a rotation of 180 degrees around a center about a line connecting the center and the geometric center of any connected atoms not in {{atomsToInvert}}. The results is the standard organic chemist\'s "stereochemical inversion" at that point -- which switches two groups on a chirality center but does not invert stereochemistry within the branches themselves. {{atomsToInvert}} is optional. If it is present, only the first atom in {{centers}} is inverted. If it is omitted, for all atoms in {{centers}}, Jmol carries out a SMARTS analysis to determine whether the center is in a ring, in which case it selects for switching the atoms that are not in the ring. If the center is not in a ring, the two shortest chains are selected for switching. See also {#.set picking~set picking invertStereo}','*12','ATOM','{{centers}}','{{atomsToInvert}}','') newCmd('','14.15.4','','*v+14.5;','Same as INVERTSELECTED ATOM {{centers}}','*11','{{centers}}','') newCmd('','12.0.RC1','','*v+12.0;','Same as INVERTSELECTED ATOM','*5','STEREO','{{centers}}','{{atomsToInvert}}','') newCmd('.isosurface','','','*v+12.0 -- adds "=XXXX" for retrieving data directly from the Uppsala electron density server, LATTICE {{a b c}} for renderning multiple copies of an isosurface, and MEP functionType to support mapping of molecular lipophilic potential;iso','
 Jmol can generate a large variety of objects using the method of isosurfaces. Many of these surfaces can be color-mapped. Two Jmol commands (isosurface and {#.mo~}) render isosurfaces. The isosurface command itself provides ultimate control over these surface renderings. Before using the isosurface command, if you are interested in rendering molecular orbitals, you should first take a look at the {#.mo~} command.
Jmol can generate a large variety of objects using the method of isosurfaces. Many of these surfaces can be color-mapped. Two Jmol commands (isosurface and {#.mo~}) render isosurfaces. The isosurface command itself provides ultimate control over these surface renderings. Before using the isosurface command, if you are interested in rendering molecular orbitals, you should first take a look at the {#.mo~} command. The general syntax of the isosurface command is as follows:
isosurface ID [object id] [construction/mapping parameters] [surface object] [additional mapping-only parameters] MAP [color mapping dataset] [display options] .
The isosurface command is complex, and while the order of parameters is flexible within these groups, parameters in different groups should not be swapped in terms of order within the command, or the result is unpredictable or will cause an error message to be displayed. While the id is optional, it is highly recommended. Specifically for the options ON, OFF, and DELETE, the id may include a wildcard character (asterisk). Thus, isosurface pl* on turns on all isosurface objects having an ID starting with "pl".
Data for these surfaces can be from several sources (see below). In addition, atom-based data from a variety of sources can be mapped onto an isosurface.
The isosurface itself represents the points in space where scalar values cross a specified "cutoff" value. Inside the surface, values are greater or equal to a specified positive cutoff or less than or equal to a specified negative cutoff. The default cutoff depends upon the type of object displayed. Note that positive and negative surfaces may be created separately, or, using the SIGN keyword, they can be generated together.
Color mapping of one object onto another is a simple as listing both an object and a dataset within the same isosurface command. Several keywords affecting the mapping are allowed. The default color scheme uses a red-->orange-->yellow-->green-->blue rainbow, where red represents minimum values and blue represents maximum values, but several other schemes are available (see below). Mapped data can be expressed as a series of contour lines.','0|1|2','','') newCmd('','','','*v+11.6;','The optional identifier allows you to refer back to this isosurface later for turning the surface on or off, deleting the surface, or changing display options. It must be either the first parameter or the second, just after DELETE. If the identifier is missing, behavior depends upon version. The ID keyword is optional but recommended, because then one can use any name for the ID; otherwise the name must not be one of the many Jmol command or keyword names. Leaving off the ID when creating an isosurface replaces the current isosurface with the new one.','TEXT','ID [object id]','') newCmd('','','','*v+11.0;','[||ADDHYDROGENS|For a solvent or sasurface object, accounts for missing hydrogens on carbon atoms only just prior to generating the surface. A simple sp3 model is used, and for that reason this option is not recommended. Instead, use {#.set_pdbaddhydrogens~set pdbAddHydrogens} to load a PDB file and automatically add hydrogen atoms to ATOM and HETATM records. ||IGNORE {{atom expression}}|Specifies a set of atoms to completely ignore when generating a solvent-related surface (SOLVENT or SASURFACE). Typically these might be solvent molecules or atoms: IGNORE {{solvent}}. Note that SASURFACE and SOLVENT surfaces by default ignore solvent molecules; VDW and MOLECULAR do not. ||IONIC radius|Atom radius relative to the ionic radius. Options include:
[|| x.x | specific absolute number of Angstroms for every atom || +/-x.x | offset greater(+) or less(-) than the ionic radius || nn% | percent of the ionic radius || +/-nn% | percent offset from the ionic radius ||] ||SELECT {{atom_exp}}|Selects a specific subset of the atoms for this surface. Same as select atom_exp; isosurface.... except the atoms are selected only within the isosurface command, not the overall script. ||SELECT {{atom_exp}} ONLY|Selects a specific subset of the atoms for this surface and also applies IGNORE {{not atom_exp}}. ||VDW radius|Atom radius relative to the van der Waals radius. Options include:
[|| x.x | specific absolute number of Angstroms for every atom || +/-x.x | offset greater(+) or less(-) than the van der Waals radius || nn% | percent of the van der Waals radius || +/-nn% | percent offset from the van der Waals radius ||] || WITHIN x.xx {{atomExpression or point}}| only creates a surface within the specified distance in Angstroms from the specified atoms or point. ||]','TEXT','[construction/mapping parameters] -- molecular/solvent surfaces','') newCmd('','','','*v+11.0;','[||COLOR color1 color2|Specifically for molecular orbitals. (for CUBE and other volume data files, use SIGN.) Specifies the two colors to use for the positive and negative lobes of a molecular orbital. Note that this and all other options in this classification must be given prior to the option that actually generates the surface.||IGNORE {{atom expression}}|Specifies a set of atoms to completely ignore when generating a molecular orbital, thus showing only selected atomic contributions.||SCALE x.xxx |In the case of molecular orbitals, scales the volume around the orbital. It may be useful in cases where Jmol misjudges the full extent of an orbital, thus truncating it.||SELECT {{atom expression}}|Selects a specific subset of the atoms for this molecular orbital. (Essentially the opposite of IGNORE.)||SIGN|For molecular orbitals derived from cube files, indicates that the data have both positive and negative values and that they should both be displayed.||SQUARED | Data are to be squared prior to surface generation. ||]','TEXT','[construction/mapping parameters] -- molecular orbitals','') newCmd('','','','*v+11.0;','[||ANISOTROPY {{a b c}}|Sets the anisotropic distortion of an object in the x, y, and z directions.||CACHE | Caches the isosurface in memory and replaces it with its JVXL equivalent. The CACHE option along with isosurface creation or alone instructs Jmol to immediately create JVXL data for the specified surface and to load that data instead. The surface remains in memory and can be used again, even after a new file is loaded using cache://isosurface_
{{cx cy cz f_ab}}|Sets the eccentricity of a sphere, ellipsoid, atomic orbital, or lobe. The first three numbers define the "principal" axis; the fourth parameter defines the ratio of the other two perpendicular axes to this main axis.||PHASE "type"|Indicates that an orbital or other object is to be colored based on the position of the voxel in space. With an atomic orbital and no parameters, indicates regions of (+) and (-) oribital value with different colors. Valid types include "x", "y", "z", "xy", "xz", "yz", "z2", and "x2-y2".||SCALE x.xxx |In the case of objects for which eccentricity can apply (spheres, ellipsoids, atomic orbitals, lobes, and volumetric data), scales the object (default 1.0).||]','TEXT','[construction/mapping parameters] -- general shapes and volumetric (cube file) data','') newCmd('','','','*v+11.8;','[||ANGSTROMS|For a cube file or user-defined function f(x,y), indicates that the volumetric definitions are in Angstroms instead of Bohr (default).||BLOCKDATA|Indicates that data for surfaces in multiple-surface CUBE files are in sequential blocks rather than the Gaussian standard of being interspersed, where all data values for a coordinate point are listed together.|| BOUNDBOX | Specifies that the surface to be generated must be within the currently defined {#.boundbox~}. || BOUNDBOX {{atomExpression or point}} {{atomExpression or point}}| Specifies that the surface to be generated must be within a volume defined by the specified two diagonal end points. || COLOR DENSITY| Allowing rendering of the actual grid of numbers (volume rendering) of the data rather than an isosurface. With CUTOFF 0.0, this setting delivers the entire set of data points. It is recommended that the BOUNDBOX parameter be used with a relatively small boundbox setting or the WITHIN parameter is used in order to not have an out-of-memory condition resulting from this option. For example: boundbox scale 1.2 {{tyr}};isosurface color density cutoff 1.6 boundbox "3hyd_map.ccp4.gz" mesh nofill If a range of values is desired, use CUTOFF [min, max] to indicate the minimum and the maximum values to be included; for example: isosurface color density cutoff [-5,10] within 4.0 {{*}} "apbs.dx". || CUTOFF x.xxx|Sets the cutoff value defining an isosurface. Typically, smaller values correspond to a larger object. Cutoffs can be either positive or negative. In the case of a molecular orbital, a positive number indicates to use both positive and negative cutoffs. Adding an explicit "+" sign before the number indicates that only the positive part of the surface is desired. (See also RMSD, below.)|| CUTOFF [min,max]|see COLOR DENSITY, above||DEBUG|Produces voluminous detail for program debugging.||DOWNSAMPLE n|"Downsampling" is the process by which an image or other data set is selectively sampled, producing a result that is grainier and has less resolution than the original data set. This option for the isosurface command indicates that only a portion of the data for a file should be used for the surface, creating an isosurface with lower resolution but with the full range of the original data. n is an integer -- downSample 2 uses only every other data point; downSample 3 takes only every third data point, etc. Supported for cube-based data sets only. ||FIXED/MODELBASED|Sets whether the surface generated is to be associated with the fixed window -- and thus appear with all frames/models -- or is to be associated with the currently displayed model (the default).||FULLPLANE| for PLANE objects, indicates that color mapping should be extended to complete the plane. || GRIDPOINTS|Adds the specific gridpoints used by the "marching cubes" algorithm for the calculation of the isosurface. Primarily for discussion and debugging of the isosurface algorithm.|| INSIDEOUT|For certain datasets it sometimes happens that the surface rendered appears inside-out (dark on the outside and bright on the inside). If this is the case, add INSIDEOUT to the isosurface command prior to specification of the file to load. Jmol will reverse the sense of what is inside and what is outside the surface. This flag only affects rendering in Jmol, not export to PovRay. ||MODEL n|Sets the identity of the model with which this isosurface is to be associated. (Defaults to the currently displayed model.)||RESOLUTION x.x|Sets the resolution of a constructed isosurface three-dimensional grid of "voxels", in points per Angstrom. Does not apply to CUBE or JVXL files, which have a resolution defined by internal file parameters.||RMSD x.x|Sets the cutoff based on the root mean square deviation of the data. (MRC/CCP4 file format; equivalent to SIGMA in Jmol 14.4; previous versions require SIGMA instead of RMSD, which is technically not a correct term.) ||SIGN c1 c2|Indicates that cube data have both positive and negative values, and that they should both be displayed using the value given for CUTOFF and its negative. The two colors to use may be given optionally.||SIGMA x.x | See RMSD, above.|| SQUARED | Data are to be squared prior to surface generation. ||SYMMETRY | Applies file-based symmetry operators to isosurface creation, resulting in more efficient creation and rendering. The default selection is {{symop=1}} ONLY, and default coloring is to color by symop based on the current setting of {#.set_propertycolorscheme~propertyColorScheme}. For example: load =1stp filter "biomolecule 1";color property symop;isosurface ID sa RESOLUTION 0.8 SYMMETRY SASURFACE 0. ||WITHIN x.xx {{atomExpression or point}}| only creates the portion of the surface within the specified distance in Angstroms of the specified atoms or point. ||WITHIN x.xx {{atomExpression or point}}| only creates a surface within the specified distance in Angstroms from the specified atoms or point. ||WITHIN -x.xx {{atomExpression or point}}| a negative value for the WITHIN distance indicates not within x.xx Angstroms of the specified atoms or point.||]','TEXT','[construction/mapping parameters] -- general file loading','') newCmd('','11.9.7','','*v+11.0;','In the case of isosurfaces with artifacts or fragments or multiple independent sets of triangles in general, options MINSET, MAXSET, SET, and SUBSET specify which sets of triangles to include. CAP and SLAB allow slicing of the isosurface based on the current boundbox or unit cell, the proximity to a point, or side of a plane. Each slab or cap creates a new fragment that can be selected or unselected using the various SET options. These commands are reversible, SET 0 specifying "all sets" again, and SLAB NONE specifying "no slabbing or capping." All of these options can be given anywhere in an ISOSURFACE command or as part of any number of additional ISOSURFACE commands. They can be strung together with no punctuation: ISOSURFACE MOLECULAR;ISOSURFACE slab none slab plane x=0 cap plane y=0 cap plane z=0. [||MINSET n
MAXSET n|Discard subsets of the surface that have at least n (MINSET) or fewer than n (MAXSET) triangles.||SET n|The SET option specifies a single set of triangles composing one specific surface fragment, where n is the 1-based set number, in decreasing order of number of triangles. ||SET 0| Reset to display all sets.||SUBSET [i,j,k,...]| Display only the specified sets, where the numbers i, j, k, ... indicate the 1-based index of the set, in order of decreasing number of triangles. So, for example, SUBSET [1] is the same as SET 1.|| ||CAP/SLAB BOUNDBOX| CAP or SLAB along the boundaries of the current boundbox. ||CAP/SLAB UNITCELL| CAP or SLAB along the boundaries of the current unit cell. ||CAP/SLAB [plane definition]| CAP or SLAB based on a standard Jmol plane description, such as "x=3" or "xy". ||SLAB within x.y {{point}}| SLAB within a give distance in Angstroms from a specified point, such as or {{1 0 0 -3}}, or an atom, such as @10 or {{protein}}. ||SLAB WITHIN RANGE x.x y.y|SLAB based on the value of a mapped parameter.||SLAB nn | SLAB dynamically at nn% of the distance from the back to the front of the isosurface. This special slab dynamically changes as the model is rotated, always slabbing based on the plane of the screen, producing a dramatic view into an isosurface regardless of the orientation of the model. ||SLAB NONE | Reset the slab, removing all results of SLAB or CAP. ||]','TEXT','[sets and slabbing options]','') newCmd('','11.9.7','','*v+11.0;','Jmol can color isosurfaces in a wide variety of ways, from simple single colors to mapped contours based on a second data set. See also the {#.colorother~color ISOSURFACE} command for additional options and changing the color of an isosurface after creation. [||COLOR <color>|Colors an isosurface the specified color name or value, such as [xFF0000]. ||COLOR "c1 c2 c3..."|Where c1, c2, etc. are color names or values, such as red or [xFF0000] within double quotes, this option allows specification of descrete colors to be used for contour mapping. ||COLOR c1 TO c2 n| (also color isosurface ......) colors an isosurface with a range of n colors from color c1 to color c2. For example, color isosurface red to white 30. ||COLOR RANGE
x.xxx y.yyy|(or color absolute) Indicates to color the specified range of value from one end to the other end of the color scheme. If numbers are not included or COLOR RANGE ALL is specified, then the minimum and maximum data values in the file are used.||COLORSCHEME "xxx" |Sets the color scheme to one of "roygb" (default rainbow), "bgyor" (reverse rainbow), "bw" (black/white), "bwr" (blue-white-red), "rwb" (red-white-blue), "wb" (white/black), "low" (red-green), or "high" (green-blue). An optional parameter TRANSLUCENT prior to the color scheme name creates a gradient of translucency from transparent to opaque across the color scheme. An additional scheme "sets" colors the isosurface different colors for different surface fragments (for example, internal cavities).||CONTOUR n|Specifies to display the object as a set of contour lines. Then number of lines is optional; 9 contour lines are shown by default. Using the CONTOUR keyword sets the default display to be CONTOURLINES NOFILL. Note that isosurface contour 0 "t.jvxl" will override contour selected in JVXL file.||CONTOUR n i|Same as CONTOUR n but draws only the ith contour line.||CONTOUR -n|With a negative number, specifies for a plane or f(x,y) object the specific single contour to depict.||CONTOUR DISCRETE
[a,b,c,d,e,...]|Sets the contour levels to discrete values. In addition, see the COLOR option, above, for the discrete coloring option.||CONTOUR INCREMENT
{{from,to,step}} |Sets the contour values starting with the from value to the to value in increments of step. In addition, see the COLOR option, above, for the discrete coloring option. ||REVERSECOLOR|For colorschemes in particular, the REVERSECOLOR option switches the direction of the color scheme. This parameter should be given just prior to the COLORSCHEME parameter.||SCALE3D x.x|generates a 3D plot of the desired scale from a mapped plane. It can be introduced either with the original definition of the isosurface or later, after the plane has been created and displayed. Negative numbers invert the graph (forming valleys instead of mountains); 0 removes the 3D scaling. Note that this rendering can be combined with CONTOUR to form a 3D "topo map". ||]','TEXT','[color and contour options]','') newCmd('','','','*v+11.0;','Several isosurface options relate specifically to molecular or solvent-related surfaces.
[||CAVITY
cr er| Renders a depiction of a molecular cavity. The optional parameters cr and er determine the overall properties of the cavity. cr (cavity radius, default 1.2) sets the radius in Angstroms of the "probe" molecule that would fit into the cavity. er (envelope radius, default 10) sets the radius of the probe used to define the outer limits of the molecule. Smaller numbers for the cavity radius lead to more detailed cavities; smaller numbers for the envelope radius lead to cavities that are more internal and extend less toward the outer edge of the molecule. Qualifier INTERIOR CAVITY creates isosurfaces only for cavities that do not extend to the outer surface of the molecule. Qualifier POCKET CAVITY creates isosurfaces only for pockets that extend to the outer surface of the model, showing them more as troughs or open "pockets". Qualifiers MINSET , MAXSET, SET and SUBSET can all be given prior to CAVITY. ||MEP|Depicts the molecular electrostatic potential, as calculated by SUM(q_i/r_i), where q_i is the partial charge on atom i (as found in the loaded model file) and r_i is the distance from atom i to a particular point in space. The molecular electrostatic potential is not typically displayed itself. Rather, it is usually mapped onto a molecular surface. For example: isosurface resolution 6 SOLVENT map MEP produces a smooth surface at the van der Waals distance around the molecule colored by the molecular electrostatic potential.||MOLECULAR|Same as SOLVENT 1.4, but solvent moleules are not ignored.||SASURFACE radius|Depicts the "solvent-accessible" surface based on the currently selected atom set. This surface is described by the center of the solvent probe as it rolls along the surface. It is larger than the "molecular" surface. The radius is optional. Solvent molecules are ignored by default. If either the VDW or the IONIC keywords are present, sasurface 0 is assumed, but solvent is not ignored.||SOLVENT radius|Depicts the "solvent-excluded" or "molecular" surface around the currently selected atoms (or the entire model if no atoms are selected). If only a subset of the atoms is selected, then only the corresponding subset of the molecular surface is depicted. This surface is defined as the surface formed by rolling a spherical solvent "probe" around the molecule at the distance of the van der Waals radii of the atoms. The specification of the radius of this probe is optional; its default setting is determined by the set radius command (Jmol default 1.2). Solvent molecules are ignored by default.||]','TEXT','[surface object] -- molecular/solvent surfaces','') newCmd('','','','*v+11.0;','Both atomic orbitals and molecular orbitals can be displayed in Jmol. Note that molecular orbitals in particular can be generated more effectively using the {#.mo~} command rather than the isosurface command. Alternatively, the "LCAO cartoon" option creates the sort of dumbbell-shaped cartoonish orbitals seen in textbooks in discussion of pi bonding and hybridization.
[|| ATOMICORBITAL
n l m Zeff|The Schroedinger solution to the hydrogen atom wavefunction. The three quantum numbers n, l, and m, must be such that abs(m) <= l < n. For solutions with imaginary roots, the two m values simply designate the two real linear combinations of these imaginary solutions. The optional effective nuclear charge, Zeff, determines the overall size of the orbital (default is 6, carbon). Add the keyword PHASE for a two-color orbital, which can be colored using the SIGN keyword followed by two color names or values.||ATOMICORBITAL
n l m Zeff POINTS nPoints radius seed|Creates a "pointilist" probability-based visualization of an atomic orbital using the specified number of points and optional radius extent and random seed. The radius parameter, if present, must be a decimal number. The integer setting {#.set_dotscale~DOTSCALE} can be increased from its default value of 1 to increase the size of the displayed points.||LCAOCARTOON
"type" (atom_exp)|Draws a lobe or p orbital (two lobes) centered at the FIRST atom of the specified expression. (Typically this would be an expression for a single, specific atom, such as atomno=3). See the {#.lcaoCartoon~lcaoCartoon} command for a discussion of the possible types of LCAO cartoons (as well as a simpler way to create them).||LOBE
{{cx cy cz f_ab}}|Draws a single tear-drop shaped object (an xy-compressed center lobe of a dz2 orbital) anywhere at any size in any direction with any amount of distortion. The first three numbers define the axis of the lobe; the fourth parameter defines its eccentricity -- the ratio of the other two perpendicular axes to this main axis.||MO n|Denotes the n-th molecular orbital described in the file that is currently loaded. Adjusting the CUTOFF to suit the situation is recommended. Molecular orbitals are automatically bicolor; color them one specific color using COLOR and just one color name or value. RESOLUTION can be used to good effect to increase or decrease the precision of the rendering. Note that only the atom-based orbitals for the currently selected atoms are utilized. Thus, one can selectively see how atomic orbitals from each atom in the molecule are contributing to any given molecular orbital.||MO HOMO/LUMO +/-n|Selects for display a molecular orbital based on energy-proximity to the highest-occupied or lowest-unoccupied molecular orbital. For example, isosurface MO HOMO or isosurface MO LUMO +3.|| MO ... POINTS nPoints seed| Produces a "pointilist" probability-based visualization of a molecular orbital using the specified number of points and an optional random seed. ||]','TEXT','[surface object] -- atomic and molecular orbitals','') newCmd('','','','*v+11.0;','There are several general shapes that can be created as "isosurfaces". These include:
[||ELLIPSOID
{{cx cy cz f_ab}}|Draws an ellipsoid having a single unique axis. The first three numbers define the "principal" axis; the fourth parameter defines the eccentricity of the ellipsoid -- the ratio of the other two perpendicular axes to this main axis.||HKL {{h k l}}|Creates a plane through a crystal based on the Miller indices hkl. Adding map molecular creates a slice through the crystal highlighting atomic positions.||PLANE|Indicates that what is desired is not really an isosurface but rather a planar slice through the data set. Using RESOLUTION 0.001 with planes allows for minimum number of triangles. Using COLOR RANGE, the range of mapped values can be changed. The range -0.008 0.008 is recommended for molecular orbitals. Planes, like other surface objects, can be mapped or left unmapped and just colored. Planes are designated using one of the methods for {#.plane expressions~}. If a BOUNDBOX keyword is given prior to PLANE (see above), then the plane will be created within that box.||SPHERE radius|Draws a sphere of the given radius in Angstroms.||]','TEXT','[surface object] -- general shapes','') newCmd('','','','*v+11.8;','Isosurfaces can be created in Jmol using external file-based "volume" or "polygon" data. Note that any of the formats that are volume data (APBS, CUBE, Chem3D, DSN6/OMAP, MRC/CCP4, XPLOR/CNS) can all be used either to generate the surface itself or (after the MAP keyword) to color a surface.
FILE "filename" n can be used in either case to depict the n-th isosurface from the specified file. Empty quotes indicate that the loaded structure file already has surface data present (CUBE files, for example, contain coordinates), and that that data should be used for construction of the surface. Thus, for example, load test.cube.gz;isosurface FILE "" will load the coordinates from the GZIPped cube file, then display its first isosurface. A default directory can be set using {#.set (misc)~set defaultDirectory}, and for applets a proxy server can be set for non-host file loading using {#.set (misc)~set appletProxy}. The keyword FILE is not required. The optional number "n" specifies which volume or surface should be used in the case of a CUBE file with multiple orbitals with multiple surfaces. All files read by Jmol may be gzip-compressed.','TEXT','[surface object] -- file-derived isosurfaces','') newCmd('','14.1.50','','*v+11.8;','Jmol can import x-ray diffraction and cyro-EM volume data maps from the {"https://www.ebi.ac.uk/pdbe/densities~European Bioinformatics Institute Density Server}, either for full structures or for localized sets of atoms. For x-ray diffraction maps, use prefixes "=" or "*" and the PDB ID, ISOSURFACE "=2gc2", for standard (2fo-fc) maps. Use "==" or "**" for difference (fo-fc) maps -- ISOSURFACE "==2gc2". (There is no difference between "=" and "*" here; "=" and "==" are legacies from when the Uppsala server was active.) If the surface is for the currently loaded PDB model, the PDB id may be omitted or the keywords eds and edsdiff may be used directly: ISOSURFACE "=" or ISOSURFACE eds. An empty string file name also is the same as eds. Default RMSD (sigma) values for x-ray diffraction maps are +1.0 for 2fo-fc maps and +/-3.0 for fo-fc maps. The difference map option automatically sets the SIGN option, returning positive and negative differences in two colors.
Cryo-electron microscopy maps are available from EBI starting with Jmol 14.31.51. To access cryo-EM maps, use the prefix "=emdb/nnnn", where nnnn is the EMDB id number. For example: ISOSURFACE "=emdb/9357" (the cryo-EM map for 6nef). A default SIGMA 10.0 will used, which is only a guess. For a specific PDB ID, use the prefix "=em/=" and the PDB ID for the EMDB id: ISOSURFACE "=em/=6nef". This queries the EMDB for the author-suggested cutoff value and uses that as the default CUTOFF. You can override that default cutoff, though, using SIGMA or CUTOFF: ISOSURFACE CUTOFF 0.03 =em/=6nef. As for x-ray volume data, use "=em/=", "=em/", or just "=em" to indicate the volume data for the current PDB model.
The DENSITY option, ISOSURFACE DENSITY, allows either cryo-EM or x-ray diffraction data to be used, first checking if cryo-EM data is available and, if not found, then checking for x-ray data. Use ISOSURFACE "=density/=xxxx", where xxxx is a PDB ID, for a model other than the current model. Which type of map was used will be indicated in the Jmol Script Console. It is also available using the GETPROPERTY ISOSURFACE command or the getproperty("isosurfaceInfo") function or, more specifically, getProperty("isosurfaceInfo.jvxlFileTitle"), which will report the type of isosurface (2FO-FC, FO-FC, or EM) as, for example, for 6NEF, "BCifDensity reader type=EM".
A default WITHIN 2.0 {{...}} isosurface option is automatically applied in all of these case. These requests use a very efficient Density Server API to retrieve just the data for the volume containing the atoms specified by WITHIN (or the full current model, if WITHIN is not specified), resulting in significant speed and bandwidth improvements. If the full set of volume data is desired -- for example, the full unit cell data for x-ray diffraction, add the suffix "/full" to the filename. For example: ISOSURFACE "=em/full" or ISOSURFACE "=em/=6nef/full".','TEXT','[surface object] -- PDB x-ray diffraction and cyro-EM surfaces','') newCmd('','','','*v+11.8;','Current volume file formats supported include:
[|| APBS | {http://apbs.sourceforge.net~APBS DX} volume data files || CASTEP | {http://www.castep.org/~CASTEP} density files||BCIF|{https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1008247~Binary CIF}|volume data maps in {https://msgpack.org~MessagePack format} served from the {https://www.ebi.ac.uk/pdbe/densities/~European Bioinformatics Institute density server}.||CUBE|{http://www.gaussian.com~Gaussian} {http://www.nersc.gov/nusers/resources/software/apps/chemistry/gaussian/g98/00000430.htm~cube} format volume data files. Units of Bohr are assumed unless the keyword ANGSTROMS precedes the FILE keyword. || Chem3D | {http://www.adeptscience.co.uk/download/dldcat/24/0/All/Chem3D.html~chem3d} 3dxml structure files may contain volume data || DSN6/OMAP (CCP4) | binary files used, for example, to deliver x-ray defraction and cryo-EM map data. ||GRASP/DELPHI| {http://www.msg.ucsf.edu/local/programs/grasp/html/Appendix%20A.html~GRASP} grid files.||Jaguar PLT | {https://www.schrodinger.com/products/14/7/~Jaguar} plt orbital files || NCI|Jmol has support for displaying results of NCIPLOT "noncovalent interactions" calculations. See "Revealing Noncovalent Interactions", Erin R. Johnson, Shahar Keinan, Paula Mori-Sanchez, Julia Contreras-Garcia, Aron J. Cohen, and Weitao Yang, J. Am. Chem. Soc., 2010, 132, 6498-6506 and "NCIPLOT: A Program for Plotting Noncovalent Interaction Regions" Julia Contreras-Garcia, Erin R. Johnson, Shahar Keinan, Robin Chaudret, Jean-Philip Piquemal, David N. Beratan, and Weitao Yang, J. of Chemical Theory and Computation, 2011, 7, 625-632. For consistency with the mapping colors of these articles, the specific color scheme shown below is recommended. Without reading any files, the NCI keyword specifies to carry out an NCI promolecular calculation. However, if the output files from {http://www.chem.duke.edu/˜yang/Software/softwareNCI.html~NCIPLOT} are available, they can be used as well, for example: ISOSURFACE parameters [0 0 0 0 0 0.01] NCI dens.cube. Note that only the density file is required; Jmol can generate the reduced density gradient data from that density file alone. Any CUBEGEN or other format density file that Jmol can read can be used here. (The 0.01 parameter is required only when reading the density file created by NCIPLOT, because that file is scaled up by a factor of 100 relative to standard density files. If using both NCIPLOT output files, then the command is simply: ISOSURFACE parameters [0.5] grad.cube MAP dens.cube fullylit.Parameters are [cutoff, p1, p2, p3, p4, p5], described below. Zero for any parameter specifies to use the default value. [||p1 | -2 to 2|0(all, default), 1(intramolecular), 2(intermolecular), -1(intramolecular NCIPLOT grad/dens filtering), -2(intermolecular NCIPLOT grad/dens filtering).||p2 |rhoMin |(min rho cutoff to remove very low density points)||p3 |rhoPlot |(cutoff used to remove covalent density, default 0.07 for promolecular, 0.05 for SCF)||p4 |rhoParam |(fraction of total rho that defines intramolecular, default 0.95)||p5 |dataScaling |(default 1, but set to 0.01 to read back in NCIPLOT -dens.cube file)||] For more information, see {misc/iso-nci.txt~}. || MRC/CCP4 | binary {http://ami.scripps.edu/software/mrctools/mrc_specification.php~AMI MAP} files have the advantage that they contain information about the root mean square deviation for the data set, allowing use of the SIGMA keyword . In some cases the keyword MRC just precede the FILE keyword in order to force MRC format reading because the file does not have the expected identifying characters M A P in the proper location.|| NFF | {http://paulbourke.net/dataformats/nff/nff1.html~neutral file format} for electron microscopy data exported from IMOD using the ISOSURFACE command || UHBD | {http://web.archive.org/web/20100701074459/http://adrik.bchs.uh.edu/uhbd.html~University of Houston Brownian Dynamics} files.|| XPLOR/CNS | {http://cci.lbl.gov/@TILDErwgk/shortcuts/htdocs/current/python/iotbx.xplor.map.html~XPLOR MAP} files ||]','TEXT','[surface object] -- volume file-derived isosurfaces','') newCmd('','','','*v+11.8;','Current surface file formats supported include:
[|| EFVET | {http://ef-site.hgc.jp/eF-site/about.html~eF-site EFVET} format surface files. Indicating a number from 0-5 after the file name specifies which type of data to map onto the surface: [||0 | color indicated in file||1 | electrostatic_potential||2 | hydrophobicity||3 | temperature_factor||4 | minimum_curvature||5 | maximum_curvature ||] || JVXL | {misc/JVXL-format.pdf~Jmol Voxel} format files are highly compressed surface files created by Jmol using the {#.write~} command. The JVXL format allows for saving the data for a selection of a specific surface cutoff from a volume file in a way that can be delivered very efficiently over the web and loaded very quickly into Jmol. || KINEMAGE | Jmol reads contact-point Kinemage files created by {http://molprobity.biochem.duke.edu~MolProbity} See {misc/kinemage.txt~} for details.|| MSMS | Jmol can read an isosurface from a set of {http://mgltools.scripps.edu/packages/MSMS~MSMS} output files. Both the .vert and the .face files must be in the same directory, or only dots will be generated. The keyword MSMS must precede the FILE keyword -- MSMS FILE "xxx.vert" -- because these files do not contain identifying information.|| OBJ | {http://autodesk.com~AutoDesk Wavefront Advanced Visualizer} {http://www.eg-models.de/formats/Format_Obj.html~OBJ} surface data files. (See also {http://www.fileformat.info/format/wavefrontobj/~Wavefront Object format.} In this case the keyword OBJ must precede the FILE keyword or the first line of the file must start with #pmesh. Jmol uses the vertex and face records of this file in order to create a set of polygons that are colored using the name of the group record, which is assumed to have the hexidecimal format g kRRGGBB. Note that Jmol does not read material (.mtl) files and so instead relies on this simpler method of assigning colors. Polygons are limited to triangles and quadrilaterals. An optional number after the file name --OBJ FILE "filename" n -- specifies a group from the OBJ file. isosurface OBJ "sample.obj" 3, for instance, reads only the third group of faces. || PMESH | Jmol can read and write both ASCII and binary JmolPmesh surface formats. This format does not allow for colored vertices and does not shade by vertex, so it is generally appropriate for polyhedra, meshes, and contours only, not filled triangulated arbitrary surfaces. In the case of an ASCII JmolPmesh file with no header line (see below), the PMESH keyword is required. Otherwise Jmol can discern the data type from the first bytes of the file contents. A "pmesh" is a surface data set consisting of a set of vertices and a set of polygons defining the "facets" of the surface. Polygons are limited to triangles and quadrilaterals. See also PMESH INLINE, below. File formats readable by Jmol include:[||numeric pmesh|This relatively simple format can be found in {misc/10x10pmesh.txt~10x10pmesh.txt}. Jmol reads this file in free format -- values simply need to be separated by some sort of white space.
#JmolPmesh\n100\n3.0000 3.0000 1.0000\n2.3333 3.0000 1.0000\n...(98 more like this)\n81\n5\n0\n10\n11\n1\n0\n...(80 more sets like this)\n
- The first line identifies the file as a Jmol Pmesh file. It is optional but recommended.
- The second line defines the number of grid points defining the surface (integer, n)
- The next n lines define the Cartesian coordinates of each of the grid points (n lines of x, y, z floating point data points)
- The next line specifies the number of polygons, m, to be drawn (81 in this case). Setting this number to -1 indicates that some number of polygons follows, terminated by a polygon with 0 points indicated.
- The next m sets of numbers define the polygons. In each set, the first number, p, specifies the number of points and format for the polygon. This number may be 0 (end of polygon list), 1 (point), 2 (edge), 3 (triangle), 4 (triangle with edges marked) or 5 (quadrilateral with edges marked) or its negative. If negative, the polygon is followed by a color written as a hexadecimal or decimal integer (0xFF00FF or 16711935). The next p numbers specify indexes into the list of data points (starting with 0). The first and last of these numbers must be identical for formats 4 and 5, in order to "close" the polygon.
\n 4 bytes: P M \\1 \\0 \n 4 bytes: (int) 1 (test for bigEndian)\n 4 bytes: (int) vertexCount\n 4 bytes: (int) polygonCount\n 64 bytes: reserved\n ---then for each vertex:\n 12 bytes: (float) x,(float) y,(float) z\n ---then for each polyhedron, \n the number of vertices (from 1 to 4) \n followed by the index of the vertex \n in the vertex list, starting with 0:\n [(int)nVertices,(int)v1,(int)v2,...,(int)vn]\n||] ||] ','TEXT','[surface object] -- surface data-derived isosurfaces','') newCmd('','','','*v+11.8;','
 Several additional sources of surfaces besides external files are available. For functionXY and functionXYZ, the extent and resolution of the grid can be set using the keywords ORIGIN, STEPS, and POINTS. For example, isosurface origin {{-5 -5 -5}} steps {{0.2 0.2 0.2}} points {{50 50 50}} functionxy = "x * y * cos(x * 100)" or isosurface cutoff 2.0 origin {{-5 -5 -5}} steps {{0.2 0.2 0.2}} points {{50 50 50}} functionxyz = "x * y * cos(z*100)" map functionxy = "x * y" (shown here on the right. [||FUNCTIONXY = "..."| graphs a function of x and y with a default range in x and y from -10 to 10 every 0.25 Angstroms. For example, isosurface functionXY = "x * y"||FUNCTIONXY
Several additional sources of surfaces besides external files are available. For functionXY and functionXYZ, the extent and resolution of the grid can be set using the keywords ORIGIN, STEPS, and POINTS. For example, isosurface origin {{-5 -5 -5}} steps {{0.2 0.2 0.2}} points {{50 50 50}} functionxy = "x * y * cos(x * 100)" or isosurface cutoff 2.0 origin {{-5 -5 -5}} steps {{0.2 0.2 0.2}} points {{50 50 50}} functionxyz = "x * y * cos(z*100)" map functionxy = "x * y" (shown here on the right. [||FUNCTIONXY = "..."| graphs a function of x and y with a default range in x and y from -10 to 10 every 0.25 Angstroms. For example, isosurface functionXY = "x * y"||FUNCTIONXY "functionName"
{{originXYZ}}
{{ni xi yi zi}}
{{nj xj yj zj}}
{{nk xk yk zk}} |The FUNCTIONXY keyword specifies that the Z-value at a given X,Y coordinate will be provided via a JavaScript function in the case of the applet or via a JmolStatusListener call in an application or, if the function name begins with file:, the numbers will be read from that file (relative to the current directory). The parameters mirror the parameters in the header of a CUBE file. Units of ANGSTROMS are assumed. Thus, we require an origin of the voxel space and for each nominal direction x, y, and z, the number of grid points and the direction of the unit vector along that edge. These four quantities must be in braces. In the case of the Jmol applet, there are three reading options. [||ni>0 and nj>0|functionName(app, ix, iy)| one point is returned for each call to the function.||ni<0 and nj>0 | functionName(app, ni, nj)|an entire set of z values separated by white space will be returned as a string. Data are read from low to high coordinates, with X changing in the outer loop (slowly) and Y changing in the inner loop: a11, a12, a13,...,a21, a22, a23,..., etc. ||ni<0 and nj<0|functionName(app, ni, nj, Fxy)| the Fxy[-ni][-nj] array will be filled with z values in the case of the applet or functionXY(functionName, ni, nj, Fxy) will be called in the case of the application. ||]
If the functionName starts with the string "file:", then Z value data will be loaded from a file instead of a JavaScript function in the order described above for reading data from a returned string.||FUNCTIONXYZ = "..."| Graphs an isosurface through a block of data defaulting to -10 to 10 Angstroms range in x, y, and z with step every 0.25 Angstroms. For example: FUNCTIONXYZ = "x * x + y + z" .|| FUNCTIONXYZ
"functionName"
{{originXYZ}}
{{ni xi yi zi}}
{{nj xj yj zj}}
{{nk xk yk zk}} | Similar to FUNCTIONXY, except that in this case the X,Y,Z cube data will be provided in the order (for x = 1 to ni)(for y = 1 to nj)(for z = 1 to nk). The first two options listed for FUNCTIONXY are not available, and the signs of ni and nj are ignored. The data structure Fxyz[ni][nj][nk] must be filled with data by the function call functionName(app, ni, nj, nk, Fxyz) in the case of the applet or functionXYZ(functionName, ni, nj, nz, Fxyz) in the case of the application.||INLINE @varName| The isosurface data may be in a variable already. For example: x = load("mydata.dat"); isosurface INLINE @x first loads the data into the variable x, then displays the isosurface from that data, possibly giving the opportunity to peek at the data first. It is advisable to reset the variable after use to improve performance, however note that the state will only be preserved if the value of the variable is left unchanged. ||PMESH INLINE "pmesh-data"| The INLINE keyword can be used with PMESH to directly create a small isosurface. Numeric pmesh data may be specified "in-line" without reference to a separate file. This is particularly useful for pmesh objects with few vertices. Note that the {#.draw~} command also can be used for this purpose.||]','TEXT','[surface object] -- additional surface sources','') newCmd('','','','*v+12.0;','If any parameters spefically relate to the mapping of the surface, not the generation of it, then they can come after the specification of the surface object. Keywords such as COLOR RANGE, CONTOUR, DEBUG, FIXED, FULLPLANE, MODELBASED, MAP, REVERSECOLOR, SCALE3D, and SELECT (when it relates to the color mapping) fall into this category. ','TEXT','[additional mapping-only parameters]','') newCmd('','','','*v+11.0;','Except for SPHERE, ELLIPSOID, LOBE, and LCAOCARTOON, which have no CUBE-file equivalent, all the other surface types (including FUNCTIONXY) can be used as CUBE-type data sets in order to color map another surface, because they all involve the intermediate generation of voxel data within Jmol. Used with PLANE as a surface object, a slice through a set of data can be color-contoured.The keyword MAP is optional, recommended for readability. The {#setisosurfacekey~set isosurfacekey} option displays a vertical color key that can be hovered over with the mouse to display the values associated with mapped colors and contours. Existing isosurfaces can be mapped or remapped. To do this, simply start the isosurface command with the MAP keyword (with optional ID). The keyword set MAP SQUARED indicates that the values should be squared prior to mapping . The Jmol parameter {#.set(misc)~isosurfacePropertySmoothing} (default TRUE) determines whether the property is smoothed out over the isourface or assigned specifically to the nearest atom.
Atom based-properties can be mapped onto a surface using one of the following options. If MEP or MEP functionType is followed by PROPERTY xxx or property_xxx or variable..., that data will be used instead of partialCharge data.
[||MEP|molecular electrostatic potential, using partial charge data within the file or assigned to atoms using the {{...}}.partialCharge = ... or {#.datasettingatomproperties~data} command syntax. A standard Coulomb function is used (1/d). ||MEP functionType| allows setting the function used for the mapping as for {http://www.symyx.com/support/developer/chime/developer_tools/chimerasmol.jsp#set_mep_mlp_function~Chime}, where functionType is the number 0, 1, 2, or 3: [|| 0|1/d|Coulomb\'s law distance function (same as rasmol potential distance function)||1|e^(-d/2)|Gaillard, P., Carrupt, P.A., Testa, B. and Boudon, A., J.Comput.Aided Mol.Des. 8, 83-96 (1994)||2|1/(1+d)|Audry, E.; Dubost, J. P.; Colleter, J. C.; Dallet, P. A new approach to structure-activity relations: the "molecular lipophilicity potential". Eur. J. Med. Chem. 1986, 21, 71-72||3|e^(-d)|Fauchere, J. L.; Quarendon, P.; Kaetterer, L. Estimating and representing hydrophobicity potential. J. Mol. Graphics 1988, 6, 203-206.||] These additional functions thus allow Jmol to use isosurface...MAP MEP to visualize molecular lipophilic potential (MLP) as well. ||PROPERTY xxx | where xxx is an atom-based property value such as partialCharge or temperature or vanderwaals. The COLOR option applies atom-based color to an isosurface, as commonly done in PyMOL. || property_xxxx | The linking underscore signifies that the property was provided separately using the DATA command and is not model-file based. A previous SELECT ...; DATA "property_xxxx"....end "property_xxxx" or, if the data are from a variable, SELECT...; DATA "property_xxxx @x", must have already been issued. Note that when data is created in this way, only the selected atoms are assigned data values. Atoms thus selected need not be contiguous, but the data will be read from the variable or DATA command line contiguously, disregarding spaces, tabs, line ends, and any string that would not evaluate to a number. This allows for targeting just a specific set of atoms for an isosurface and associated data. After the property name the keyword WITHIN and a distance in Angstroms can be added to speed the calculation. The default for set isosurfacePropertySmoothing FALSE is 5.0 Angstroms. ||VARIABLE x| The property is in a varible named "x", possibly from a command such as x = load("mydata.txt"). In this case, the variable must contain a value for every atom in the model, even if only a subset of the atoms is being used for the surface. ||] ','TEXT','MAP [color mapping dataset]','') newCmd('','','','*v+11.0;','Display options are indicated in the following table. These may be given at the end of the definition of the surface or in a later command having just these keywords and the identifier (or "ALL") of the desired isosurface.
[||BACKSHELL/NOBACKSHELL|Opposite of FRONTONLY; displays only the back of the isosurface.||CONTOURLINES/NOCONTOURLINES| turns on and off contour lines generated using the CONTOUR option (see bove). ||DOTS/NODOTS| Display dots at each mesh vertex point. The integer setting {#.set_dotscale~DOTSCALE} can be increased from its default value of 1 to increase the size of these dots. ||FILL/NOFILL| Display the isosurface as a smoothly filled surface.||FRONTONLY/NOTFRONTONLY| Display only the front half of the surface. This can be useful when the options mesh nofill are used. ||FRONTLIT /BACKLIT /FULLYLIT| In some cases the isosurface may appear flat. Using BACKLIT will fix this; FULLYLIT makes exterior and interior surfaces bright; FRONTLIT is the default setting. ||MESH/NOMESH| Display a mesh of lines intersecting at the vertexes used to construct the isosurface. ||OFFSET {{x y z}}| Offset the isosurface by the specified absolute amount. ||ROTATE quaternion/NONE| Rotate the isosurface by a given amount around a given axis. Rotations are about {{0 0 0}} and are cumulative; ROTATE NONE returns the isosurface to its original orientation. For example: isosurface s1 ROTATE @{{quaternion({{1 0 0}}, 90)}} ||ON/OFF| Turn the isosurface ON or OFF, but do not delete it. ||OPAQUE/TRANSLUCENT n| Display the isosurface as an opaque or translucent object. Several translucent options are available; see the {#.color~} command. ||TRIANGLES/NOTRIANGLES| Display separated triangles (primarily for debugging purposes).||]','TEXT','[display options] ','') newCmd('','','','*v+11.8;','Calculates the area of the current isosurface in �2 and stores that value in the isosurfaceArea variable. The AREA parameter may also accompany any other parameters in the construction of an isosurface. The result is an array unless the optional specific surface set has been specified., in which case it is a decimal number. Sets are ordered from largest to smallest. SET 0 gives the sum of all areas. SET -1 returns the calculation to report the full array.','*21','AREA','SET','(integer)','') newCmd('','12.1.48','','*v+12.2;','Connects an isosurface to a specific reference atom so that if that atom is moved (using {#.rotateSelected~}, for instance) or hidden the isosurface moves or is hidden with it. Typically the atom expression is a model, for example {{2.1}}, but only the first atom in the atom expression is used. Use isosurface connect {{none}} to disconnect an isosurface from its atom. Note that if more than one model (frame) is visible and this surface is for a specific model only, it is still necessary to add both the isosurface MODEL and SELECT options: frame *;isosurface model 1.2 connect {{1.2}} select all vdw.','*22','CONNECT','._atom_expression') newCmd('','','','*v+11.0;','Deletes all isosurfaces. ','*31','DELETE','') newCmd('','12.0.RC9','','*v+12.0;','You can duplicate isosurface areas based on the unit cell lattice. This is a rendering option, so it can be applied any time after an isosurface is created. It is best done with packed unit cells and slabbed isosurfaces. For example: load quartz.cif {{1 1 1}}; isosurface slab unitcell vdw; isosurface lattice {{3 3 3}} creates an isosurface that spans nine unit cells. The LATTICE keyword may also appear after the MAP keyword, indicating that the mapped data is periodic and should be mapped for multiple unit cells.','*32','LATTICE','{{a b c}}') newCmd('','14.3.10','','*v+14.4;','Creates an isosurface from periodic volumetric data with the specified number of unit cells. The FIXED option "fixes" the isosurface in the sense that the operation is at load time not at rendering, allowing slabbing and use of WITHIN. You can duplicate isosurface areas based on the unit cell lattice. This is a rendering option, so it can be applied any time after an isosurface is created. It is best done with packed unit cells and slabbed isosurfaces. For example: load quartz.cif {{1 1 1}}; isosurface slab unitcell vdw; isosurface lattice {{3 3 3}} creates an isosurface that spans nine unit cells. The LATTICE keyword may also appear after the MAP keyword, indicating that the mapped data is periodic and should be mapped for multiple unit cells.','*33','LATTICE','{{a b c}}','FIXED','') newCmd('','11.1.28','','*v+11.2;','Lists all isosurfaces','*39','LIST','') newCmd('','','','*v+11.8;','When an isosurface consists of more than one set of surfaces (perhaps from isosurface internal cavity), specifying a SET displays just that single subset of the overall isosurface. SET 0 returns the display to all sets. Sets are ordered from largest (1) to smallest.','*400','SET','(integer)') newCmd('','14.31.15','','*v+11.8;','Similar to ISOSURFACE SET but displays one or more subsets of the overall isosurface. For example, ISOSURFACE SUBSET [1 2 3] will display the three largest sets. ','*405','SUBSET','(array)') newCmd('','12.1.50','','*v+12.2;','Isosurface SLAB provides an extensive mechanism allowing for the trimming of an isosurface using a wide variety of methods. Isosurfaces can be "slabbed" based on planes, spheres, bounding boxes, unit cells, proximity to a set of atoms, a range of mapped values, or a dynamic z-plane relative to the user (as in Rasmol slabbing of models, but with isosurfaces instead). The section removed can be removed completely or, using isosurface SLAB TRANSLUCENT, left as a translucent "ghost" surface. An isosurface may be slabbed any number of times in any number of ways. The slabbing is reversible using isosurface SLAB NONE for general slabbing and isosurface SLAB OFF for dynamic z-plane slabbing. Options include: [|| {#.[plane expression]~(plane expression)} | a plane. Note that plane definitions starting with "-" switch sides of the plane ||BOUNDBOX | within the currently defined {#.boundbox~bounding box} ||UNITCELL | within the currently defined {#.unitcell~unit cell} ||WITHIN x.y {{atom expression}} | within the given distance of the specified atom set ||WITHIN x.y (point) | within the given distance of the specified point (a spherical slab) ||WITHIN RANGE x.x y.y | within the mapped value range x.x and y.y. ||]','*411','SLAB','(parameters)') newCmd('','14.3.10','','*v+14.4;','Isosurface UNITCELL x.x is for periodic lattices only. It adjusting the grid by x.x in fractional coordinates. Caution is advised, as the grid is expanded in this process, leading to more grid points and more memory required. Negative x.x results in a selection of a subset of the data centered on the center of the unit cell.','*412','UNITCELL','(fraction)') newCmd('','','','*v+11.8;','Calculates the volume of the current isosurface in cubic Angstroms and stores that value in the isosurfaceVolume variable. The VOLUME parameter may also accompany any other parameters in the construction of an isosurface. The result is an array unless the optional specific surface set has been specified., in which case it is a decimal number. Sets are ordered from largest to smallest. SET 0 gives the sum of all volumes. SET -1 returns the calculation to report the full array.','*42','VOLUME','SET','(integer)','') newCmd('.javascript','11.1.28','','*v+11.2 -- new;script;javascript','Executes the specified javascript command. Applet only; may be disallowed by setting Info.allowJavaScript = false for the Info structure used when creating the applet. Note that double quotes or no quotes are required; single quotes are not appropriate in this context.','1|2','"javascript commands"','') newCmd('.label','','','*v+11.8 -- extends atom options with %[...];label;latom','Turns on and off atom labels based on a previous selection. If a string is given, it is used as the label. See also {#.hover~} and {#.echo~}. Additional options include setting the {#.font~}, {#.color~color}, {#.background~background}, x- and y-{#.set_labeloffset~offsets}, and the text {#.set_labelalignment~alignment} (left, center, or right). Default settings for these characteristics can be set by first issuing select none, so that no real label is set. Note that commands such as cartoons ONLY with the ONLY keyword turn off labels. To use ONLY and preserve labels, simply assign a variable to the labels to be kept and then restore them -- for example, Var x = {{*}}.labels; cartoons ONLY; {{*}}.labels = x.','1|2','','') newCmd('','','','*v+11.8;','You can specify atom parameters two different ways. You can use the older notation consisting of a percent sign followed by a single character, or you can use a new notation that is more flexible and easier to remember consisting of a percent sign followed by a keyword in brackets. For instance, you can use select *.ca;label "%n%R" or select *.ca;label "%[group]%[resno]". See {#.atomproperties~atom properties} for a detailed list of atom properties that can be included in labels. In addition, you can read string data into labels from a {#.data~data "mydata"} command using label %{{mydata}}. If data of name "mydata" is not found, the model\'s auxiliaryInfo will be checked. For example, with P2N files, which add "altName" information for an atom, one can then use label %{{altName}} to label atoms using that alternative name.','TEXT','Atom label parameters','') newCmd('','','','*v+11.8;','The sign of the digit x in %x.y determines the column formatting, and the digit y determines decimal places for decimal numbers or maximum number of characters for strings. In addition, if y is negative, for numbers, the number is expressed in scientific notation, and for strings, the string is truncated from the right instead of from the left. In the table below are two examples, one numeric and one a string. Leading and trailing periods represent space characters. [|| %x.y | 123.4 | "tetrahedral" || %10.3 | ...123.400| .......tet || %-10.3 | 123.400... | tet....... || %10.-3 | ..1.234E+2 | .......ral || %-10.-3 | 1.234E+2.. | ral....... ||] Note that both x and y are independently optional. %6[temperature] would be the unrounded temperature value placed in a 6-character field (and allowed to run over); %.3[shape] would be the first three letters of the the shape of the atom. ','TEXT','Column formatting and scientific notation','') newCmd('','','','*v+11.8;','You can use labels for more than just labeling atoms. Using the .label() function you can create variables and save them to files or deliver them to a webpage using JavaScript, thus producing customized output of your choice. For example, the commands
x = {{*.ca}}.label("%5[group] %7[resno] %10.2phi %10.2psi");
write VAR x "phipsi.xls"
creates a file that lists phi and psi Ramachandran angles for a protein. (A similar output can be obtained using the {#.write~write Ramachandran} command.) From a web page, the following JavaScript retrieves bond information for the first silicon atom from an applet:
var x = jmolEvaluate(\'print {{_Si}}[1].bonds.label("%6a1 %6a2 %ORDER %4.2LENGTH")\')
Note that the {#.write~} command can be used with the signed applet from the {#.console~} to save data on a local drive even if the applet is from another source, such as {http://www.rcsb.org~}. ','TEXT','Label applications','') newCmd('','','','','Each label is associated with a specific atom center (a label is an atom property). Labels for all atoms that are {#.select~selected} are affected by any label command. You can position a label relative to the center of its associated atom using -{#.set_labeloffset~set labelOffset}, and align the text using {#.set_labelalignment~set labelAlignment} (LEFT, CENTER, or RIGHT). If you want to position text at the average position of a group of atoms (or drawn objects, or an arbitrary x,y,z position), use {#.echo~} instead. For example: set echo pt1 {{20.0 30.0 23.0}};echo "HERE"; or set echo group1 {{[ala]120}};echo ALA;.','TEXT','Label positioning','') newCmd('','','','*v+11.8;','The basic commands for turning labels on and off are label xxx, where xxx is the text or format for the label, label ON, label OFF, label DISPLAY, and label HIDE. Setting a label to the empty string "" turns it off. In addition, the following SELECT commands allow selection of atoms based on their labels:
select label == ""
select label != ""
select label = "N" (not case specific)
select label like "A*" (case specific; allows wildcard)
See also {#.set_LabelToggle~set labelToggle}. ','TEXT','Setting Labels and Turning Them On and Off','') newCmd('','','','*v+11.8;','Labels on delivers a default label to the currently selected atoms. The default for this label (%[identify]) depends upon the file type and whether more than one file is loaded:
[|| single model, non-PDB data| %[atomName] #%[atomNo]||mutiple model, non-PDB data| %[atomName]/%[model #%[atomNo]. Note that the model will be displayed in "file.model" notation. For example, C12/3.2 #10 would be an atom in the third model of the second file loaded.|| single model, PDB data| %[group]%[sequence]:%[chain].%[atomName]%%%[altloc] #%[atomNo]. (Not all residues will show the chain or altloc, in which case the preceding colon or percent sign, respectively, is not displayed. For example: [MET]1:C.N #608.)||mutiple model, PDB data| %[group]%[sequence]:%[chain].%[atomName]%%%[altloc]/%[model] #%[atomNo]||]
Note that labels off deletes the current formatting, and labels on always restores the default label. ','2','._on_off{"ON"}','') newCmd('','','','','The label command followed by the desired text in quotes creates and displays that label for the currently selected atoms. The quotes are recommended but not necessary if no special characters such as semicolon, braces, or pound sign are present. Use | (vertical bar) or \\n as a line separator for multiline labels. A limited amount of HTML-like formatting can be used: [||<sub> </sub>|subscript||<sup> </sup>|superscript||<color COLOR> </color>|font color; for example label "%i <color red>%a</color> or label "%i <color #ff0000>%a</color>". ||]Single quotes do not count as quotes in this context, but the text may be a simple variable: x = "testing";label @x.','1','"text"','') newCmd('','','','*v+11.8;','Labels DISPLAY ensures that a label is displayed, creating a new default label if need be.','3','DISPLAY','') newCmd('','','','*v+11.8;','Labels HIDE hides labels for the currently selected atoms without deleting them or changing their text.','4','HIDE','') newCmd('.lcaoCartoon','','','*v+11.4;iso','
 The lcaoCartoon command displays cartoonish atomic p and hybrid sp, sp2, sp3, sp3d, and sp3d2 orbitals like those commonly seen in textbooks in discussions of the method of linear combination of atomic orbitals.
The lcaoCartoon command displays cartoonish atomic p and hybrid sp, sp2, sp3, sp3d, and sp3d2 orbitals like those commonly seen in textbooks in discussions of the method of linear combination of atomic orbitals. Any number of the following options may be strung together in the same command. The {#.isosurface~} command can also be used for the creation of LCAO cartoons. Selections for scale, color, and translucency are "persistent" -- carrying over from one command to the next -- and thus need only to be given once per model loading if they are not to be changed. Note that these options must be given prior to or along with the actual command creating the cartoon; they do not act on lcaocartoons that have already been made. The CREATE keyword and its associated orbital type, optionally with the added keyword MOLECULAR, must be the very last keyword when multiple keywords are involved.','1|2','','') newCmd('','','','*v+11.0;','Turn on/off the selected LCAO cartoon.','*1','._on_off{"ON"}','') newCmd('','','','*v+11.0;','Creates a new LCAO cartoon (a tear-shaped lobe) of the given type at the currently selected atoms. Of the selected atoms, only atoms with compatible sigma hybridization are used. The CREATE keyword is optional. Other options include LONEPAIR (two small dots) and RADICAL (one small dot) instead of CREATE . Valid types include are shown below. In addition to those listed, adding a "-" sign prior to the designation -- "-sp2" for example -- denotes the position 180 degrees rotated from the described position. For sp2, sp3, sp3d, and sp3d2 hybridizations, the order a-f starts with bonds present in the model in order of atomIndex and continues to unoccupied locations. (Note: Previous versions of Jmol were inconsistent in terms of this ordering.)
[||"cpk"| Creates a sphere at the current spacefill radius. The reason this could be useful is that such spheres, though associated with an atom, can be slabbed and capped like an isosurface. This allows for a useful "unit cell only" rendering of spacefill models, for example, using spacefill ionic;lcaocartoon scale 1.0 CAP unitcell "cpk";spacefill off. (We turn the spacefill off -- it was just to provide the reference sizes.)||"lp"|a lobe at the "lone pair" position at an AX3E center such as the N atom in NH3, with three bonds in sp3 sigma hybridization, or the sp2-hybridized lone pair in an AX2E bent center such as the unprotonated N atom in histidine. (For two dots, use LONEPAIR instead of CREATE.) ||"lpa", "lpb"|a lobe at one of the two sp3 sigma-hybridized lone pair positions at an AX2E2 bent center, such as that in H2O. (For two dots, use LONEPAIR instead of CREATE.)||"px" "py" "pz"|Standard p orbitals at an sp or sp2 sigma-hybridized center. Both lobes "a" and "b" are produced unless LONEPAIR or RADICAL is being used instead of CREATE.||"s"|standard spherical s orbital at any center.||"spa" "spb"|the two sigma bonding/nonbonding positions around a linear sp sigma-hybridized center.||"sp2a" "sp2b" "sp2c"|the three sigma bonding/nonbonding positions around a trigonal planar or bent center.||"sp3a" "sp3b" "sp3c" "sp3d"|the four sp3 sigma-hybridized positions at any center with a geometry consistent with a tetrahedral electron configuration (bent, trigonal pyramidal, tetrahedral).||"sp3da" "sp3db" "sp3dc" "sp3dd" "sp3de"|the five sp3d sigma-hybridized positions at any center that has at least three bonds and is consistent with a trigonal bipyramidal electron configuration (for example, T-shaped or see-saw). ||"sp3d2a" "sp3d2b" "sp3d2c" "sp3d2d" "sp3d2e" "sp3d2f"|the six sp3d2 sigma-hybridized positions at any center that has at least three bonds and is consistent with an octahedral electron configuration. ||]','*20','CREATE','"[type]"') newCmd('','11.8.RC4','','*v+11.8;','Sets two small dots instead of a lobe at the given position.','*21','LONEPAIR','"[type]"') newCmd('','11.8.RC4','','*v+11.8;','Sets one small dot instead of a lobe at the given position.','*22','RADICAL','"[type]"') newCmd('','','','*v+11.0;','Creates a new LCAO cartoon of the given type using the molecular axes system as reference. Applicable only for "px", "py", "pz", "-px", "-py", and "-pz". ','*3','CREATE','"[type]"','MOLECULAR','') newCmd('','','','*v+11.0;','Colors the orbital one specific color for the lcaocartoon being created with that command or future lcaocartoons.','*4','COLOR','._colorRGB') newCmd('','11.1.38','','*v+11.2;','Allows for translucent or opaque lobes for the lcaocartoon being created with that command or future lcaocartoons. For translucency options, see {#.color~}.','*9','TRANSLUCENT or OPAQUE','') newCmd('','','','*v+11.0;','If this is a p orbital, colors the two lobes different colors for the lcaocartoon being created with that command or future lcaocartoons.','*60','COLOR','._colorRGB','._colorRGB','') newCmd('','','','*v+11.0;','Delete the LCAO cartoons on the currently selected set of atoms.','*62','DELETE','') newCmd('','11.1.28','','*v+11.2;','Lists all LCAO cartoons','*65','LIST','') newCmd('','','','*v+11.0;','Sets the scale of the LCAO cartoon for the lcaocartoon being created with that command or future lcaocartoons.','*7','SCALE','(decimal)') newCmd('','','','*v+11.0;','For the already selected atom set, selects what type of orbital for an on/off/delete operation.','*82','SELECT','"[type]"') newCmd('','11.1.38','','*v+11.2;','Selects a set of atoms. In the absence of this keyword, the previously selected atom set is used.','*81','SELECT','._atom_expression') newCmd('.[read/write ZIP files]','11.3.48','','*v+11.4;','Jmol can read and write ZIP file collections. In any file reading operation (for example, in the {#.isosurface~}, {#.load~}, or {#.script~} command), Jmol can read a specific file within ZIP or JAR file collections. (The two formats are the same.) Simply indicate the path to the file within the collection using the vertical bar character as a separator: load "myfiles.zip|files|pdb|1crn.pdb". A path element may be a subdirectory within the ZIP file or the name of a ZIP file contained within one of those directories: isosurface "mysurfaces.zip|isousurfaces|jvxl.zip|surface1.jvxl". The full directory contents of a ZIP or JAR file can be read out using getProperty fileContents "myfile.zip", and the contents of specific files within a zip collection can also be read this way as, for example, getProperty fileContents "myfile.zip|JmolManifest". Note that variables can be assigned these contents using the getProperty() function: var dir = getProperty("fileContents", "myfile.zip|JmolManifest"). The {#.load~} command specifically allows reading of multiple files from a ZIP or JAR collection, and will read files in the order specified in a file with the name "JmolManifest" contained in the compressed file collection. The command {#.write~write xxx.jmol} or {#.write~write ZIPALL xxx.zip} will create a ZIP file collection that contains a JmolManifest file along with all of the files necessary to recreate the current state. In addition, {#.write~write ZIP xxx.zip} will make a collection that contains all local files necessary, but will simply point to any required remote (http://, ftp://) resource.','1|2','','') newCmd('.load','','','*v+13.0 adds capability to load from PubChem and to read JCAMP-DX (JDX) files (IR, NMR, UV/VIS, GC/MS) and to correlate atoms with peak assignments or molecules and MS fragments;load;lload','Loads the specified file or URL or string data. A wide variety of file types are supported. In general, resolution of file type is based on internal file cues, not the filename or file extension. However, this resolution process can be overridden by specifying a prefix to the file name consisting of the file type followed by two colon characters: load "molpro(xml)::myfile". PDB-type files can be loaded with automatic addition of hydrogen atoms and multiple bonds using {#.set_pdbaddhydrogens~SET pdbAddHydrogens}. For PDB, PQR, and mmCIF files that contain HELIX, SHEET, or TURN records provided by the author, those structures are created (though not initially displayed); for PDB, PQR, and mmCIF files that do not contain this information, Jmol will generate secondary structure using DSSP (W. Kabsch and C. Sander, Biopolymers, vol 22, 1983, pp 2577-2637; {http://swift.cmbi.ru.nl/gv/dssp~}) for chains that contain backbone atoms and the method of Levitt and Greer [J.Mol.Biol.(1977) 114, 181-293] for alpha-carbon-only chains. (See also {#.calculate~calculate STRUCTURE}.)
Multiple files and a selected model from a multi-model file can be read as well. Files may be Gzipped, and multiple files can be read from compressed ZIP and JAR collections. A default directory can be set using {#.set_defaultDirectory~}, and for applets a proxy server can be set for non-host file loading using {#.set_appletProxy~}. After the filename, the options listed below are available. These options must appear in the order given in the table below. In addition, the load command can take parameters that specify the number of unit cells to generate, the the space group or Jones-Faithful operators to use, and the unit cell dimensions. Using these parameters, the space group and unit cell information within a file can be overridded, and even simple XYZ data files can be turned into crystallographic data sets. The FILTER keyword allows selective loading of atoms as well as construction of the biologically relevant molecule (PDB "BIOMOLECULE" records. See also the headings {#.load APPEND~}, {#.load FILES~}, {#.load MENU~}, {#.load MODELS~}, and {#.load TRAJECTORY~}. Note that with the Jmol application (not the applet) you can also use Edit...Paste to load molecular coordinate from the system clipboard. The same capability for the applet can be had using {#.data~data "model"}. ','1|2','','') newCmd('.load','','','*v+12.2 adds the SUPERCELL option and automatic addition of hydrogen atoms and multiple bonds to PDB files;*v-13.0;load','Loads the specified file or URL. A wide variety of file types are supported. In general, resolution of file type is based on internal file cues, not the filename or file extension. However, this resolution process can be overridden by specifying a prefix to the file name consisting of the file type followed by two colon characters: load "molpro(xml)::myfile". PDB-type files can be loaded with automatic addition of hydrogen atoms and multiple bonds using {#.set_pdbaddhydrogens~SET pdbAddHydrogens}. For PDB, PQR, and mmCIF files that contain HELIX, SHEET, or TURN records provided by the author, those structures are created (though not initially displayed); for PDB, PQR, and mmCIF files that do not contain this information, Jmol will generate secondary structure using DSSP (W. Kabsch and C. Sander, Biopolymers, vol 22, 1983, pp 2577-2637; {http://swift.cmbi.ru.nl/gv/dssp~}) for chains that contain backbone atoms and the method of Levitt and Greer [J.Mol.Biol.(1977) 114, 181-293] for alpha-carbon-only chains. (See also {#.calculate~calculate STRUCTURE}.)
Multiple files and a selected model from a multi-model file can be read as well. Files may be Gzipped, and multiple files can be read from compressed ZIP and JAR collections. A default directory can be set using {#.set_defaultDirectory~}, and for applets a proxy server can be set for non-host file loading using {#.set_appletProxy~}. After the filename, the options listed below are available. These options must appear in the order given in the table below. In addition, the load command can take parameters that specify the number of unit cells to generate, the the space group or Jones-Faithful operators to use, and the unit cell dimensions. Using these parameters, the space group and unit cell information within a file can be overridded, and even simple XYZ data files can be turned into crystallographic data sets. The FILTER keyword allows selective loading of atoms as well as construction of the biologically relevant molecule (PDB "BIOMOLECULE" records. See also the headings {#.load APPEND~}, {#.load FILES~}, {#.load MENU~}, {#.load MODELS~}, and {#.load TRAJECTORY~}. Note that with the Jmol application (not the applet) you can also use Edit...Paste to load molecular coordinate from the system clipboard. The same capability for the applet can be had using {#.data~data "model"}. ','1|2','','') newCmd('.load','','','*v+12.0 adds SMILES string reading;*v-12.2;load','Loads the specified file or URL. A wide variety of file types are supported. In general, resolution of file type is based on internal file cues, not the filename or file extension. However, this resolution process can be overridden by specifying a prefix to the file name consisting of the file type followed by two colon characters: load "molpro(xml)::myfile".
Multiple files and a selected model from a multi-model file can be read as well. Files may be Gzipped, and multiple files can be read from compressed ZIP and JAR collections. A default directory can be set using {#.set_defaultDirectory~}, and for applets a proxy server can be set for non-host file loading using {#.set_appletProxy~}. After the filename, the options listed below are available. These options must appear in the order given in the table below. In addition, the load command can take parameters that specify the number of unit cells to generate, the the space group or Jones-Faithful operators to use, and the unit cell dimensions. Using these parameters, the space group and unit cell information within a file can be overridded, and even simple XYZ data files can be turned into crystallographic data sets. The FILTER keyword allows selective loading of atoms as well as construction of the biologically relevant molecule (PDB "BIOMOLECULE" records. See also the headings {#.load APPEND~}, {#.load FILES~}, {#.load MENU~}, {#.load MODELS~}, and {#.load TRAJECTORY~}. Note that with the Jmol application (not the applet) you can also use Edit...Paste to load molecular coordinate from the system clipboard. The same capability for the applet can be had using {#.data~data "model"}. ','1|2','','') newCmd('','14.1.16','','*v+11.6;','Jmol reads a wide variety of data formats. Example files can be found at {http://jmol.svn.sourceforge.net/viewvc/jmol/trunk/Jmol-datafiles~}. Supported file types include:
[||$identifier|Jmol can utilized the SMILES-to-3D and database service at the {https://cactus.nci.nih.gov~NIH Cactus server} to load 3D models based simply on {#.Jmol SMARTS/SMILES~SMILES} strings or chemical identifiers such as chemical names, CAS registry numbers, and InChI keys. For example, load $CCC loads the structure of propane, load $C1CCCCC1 loads a chair cyclohexane model, and load $tylenol loads a model of acetaminophen. || :name|The signed applet and application can load a model directly from PubChem. A name or a PubChem ID may be given. For example, load ":tylenol" or load ":1983" loads a model of acetaminophen. More specifically the syntax load :xxx:yyyy, where xxx is CAS, CID, NAME, INCHI or SMILES, specifies the exact sort of compound identifier yyyy that follows the second colon. Note that INCHI must be a standard inchi key, as perhaps generated using {{*}}.find("chemical", "stdinchikey")||=XXXX|a PDB file directly from RCSB. For example, load "=1crn".||=XXXX.cif|a CIF file directly from RCSB. ||=XXXX.mmtf|an MMTF file directly from RCSB. The {https://mmtf.rcsb.org~The Macromolecular Transmission Format} is a new compact binary format to transmit and store biomolecular structural data quickly and accurately. ||==XXX|a ligand from RCSB. For example, load ==hem.||=XXXX/dssr|Adding /DSSR instructs Jmol to also obtain the nucleic acid secondary structure {http://x3dna.bio.columbia.edu/~3DNA} from Columbia University. For a full discussion of Jmol in relation to DSSR see {https://doi.org/10.1093/nar/gkx365~Robert M. Hanson, Xiang-Jun Lu, DSSR-enhanced visualization of nucleic acid structures in Jmol, Nucleic Acids Research, Volume 45, Issue W1, 3 July 2017, Pages W528-W533}.||=XXXX/rna3d|loads mmCIF data from RCSB, also fetching RNA secondary structure annotations from {http://rna.bgsu.edu/rna3dhub~} indicating hairpinLoops, internalLoops, and junctions. Allows for such constructs as
select within(rna3d,"hairpinLoops") :> select within(rna3d,"hairpinLoops where index=5"):> x = getproperty("auxiliaryInfo.models[1].rna3d.internalLoops.5"):> x = getproperty("auxiliaryInfo.models[1].rna3d.internalLoops")[5]:> print x:> {{:> _atoms : ({{3023:3062 3639:3706}}):> _isres : true:> _path : "internalLoops":> index : 5:> units : "1S72¦1¦0¦C¦153,1S72¦1¦0¦C¦154,1S72¦1¦0¦G¦182,1S72¦1¦0¦A¦183,1S72¦1¦0¦G¦184":> }}:> y = x._atoms:> select y||=XXXX/dom | see *XXXX/dom; mmCIF data retrieved from RCSB||=XXXX/val | see *XXXX/val; mmCIF data retrieved from RCSB|| *XXXX | Similar to =XXXX, but retrieving data from {http://www.ebi.ac.uk/pdbe~}. || *XXXX/dssr | see =XXXX/dssr; mmCIF data retrieved from PDBe|| *XXXX/rna3d | see =XXXX/rna3d; mmCIF data retrieved from PDBe|| *XXXX/dom | mmCIF + domain annotations from PDBe.|| *XXXX/val | mmCIF + validation annotations from PDBe.|| ADF|{http://www.scm.com/~Amsterdam Density Functional} output file||=ams/nnnnnnn | (seven-digit number) loads data from American Mineralogist Crystal Structure Database, fetching data from http://rruff.geo.arizona.edu/AMS; employs "#_DOCACHE_" flag, indicating to save the full structure in a state file, because this number is not a permanent reference in the database.For example: LOAD =ams/0014673 same as LOAD http://rruff.geo.arizona.edu/AMS/viewJmol.php?amcsd=14673&action=showcif#_DOCACHE_||=ams/nnnnn | (five-digit number) loads that specific structure based on sequential id code.For example: LOAD =ams/10000 same as LOAD http://rruff.geo.arizona.edu/AMS/viewJmol.php?id=10000&action=showcif#_DOCACHE_||=ams/mineralName | loads all AMS structures for a specified named mineral. For example: LOAD =ams/diamond same as LOAD http://rruff.geo.arizona.edu/AMS/viewJmol.php?mineral=diamond&action=showcif#_DOCACHE_||=chebi/nnnnnn|{https://www.ebi.ac.uk/chebi~Chemical Entities of Biological Interest} 2D model with minimal 100-step minimization||=cod/number|For example, load =cod/1000041. Loads {http://www.crystallography.net~Crystallographic Open Database} files by number||AFLOW|{http://www.aflowlib.org~AFLOW} binary alloy files. ||AIMS|{https://aimsclub.fhi-berlin.mpg.de/~Ab Initio Molecular Simulation Package} (input files only) ||AMPAC|{http://www.semichem.com/ampac/~AMPAC} file ||Argus(XML)|{http://www.planaria-software.com/~ArgusLab} AGL file||Bilbao(BCS)|{http://cryst.ehu.es~Bilbao Crystallographic Server} file format. This simple format is similar to XYZ file format, but including International Tables space group number (first line), the unit cell parameters (second line), an atom count (third line), and then a list of atom names and positions. ({http://sourceforge.net/p/jmol/code/HEAD/tree/trunk/Jmol-datafiles/bcs/~examples})||CGD|{http://gavrog.org/Systre-Help.html#file_formats~Gavrog Systre} Crystal Graph Data file format, as produced by {http://www.topos.samsu.ru~TOPOS} topological analysis of crystal structures.||Chem3D(XML)|CambridgeSoft {http://www.cambridgesoft.com/software/details/?ds=3~Chem3D} C3XML file||CASTEP|{http://www.castep.org~CASTEP} input, .md, .geom, .phonon, .magres, and .ts file formats. .phonon files are read as trajectories -- see the FILTER specifications, below, to load specific q-point mode sets.||CIF|International Union of Crystallography {http://www.iucr.org/iucr-top/cif/home.html~Crystallographic Information File}, versions CIF 1.1 and CIF 2.0, including Macromolecular Crystallographic Information files (mmCIF), Modulated Structure files (msCIF), and Bilbao Magnetic structure files ({http://158.227.5.82/magndata/~mCIF}). NOTE: Jmol 12.2 changes the default for loading CIF files that have GEOM_BOND records to make loading more similar to the way Mercury opens files. Such files are now loaded as full molecules based on the idea that a "molecule" is "symop=1 or connected(symop=1)". The model is displayed WITHOUT a unit cell, and no unit cell information is made available. The standard load xxx.cif {{1 1 1}} is still available if a unit cell is desired.||CML(XML)|{http://cml.sourceforge.net/~Chemical Markup Language} files. ||CRYSTAL| {http://www.crystal.unito.it/~Crystal09} solid state computation output, including support for 1D (polymer) and 2D (slab) periodicity. This file format creates the atom properties property_spin and property_magneticMoment . ||CSF|Fujitsu {http://www.computers.us.fujitsu.com/www/products_bioscience.shtml?products/bioscience/cache~Sygress Explorer} (formerly CAChe) chemical structure file, including the reading of ab initio, semiemperical, gaussian, and density functional molecular orbitals||CUBE|Gaussian {http://www.gaussian.com/g_ur/u_cubegen.htm~cubegen} output file||DCD| single precision binary (FORTRAN unformatted) {http://www.ks.uiuc.edu/Research/vmd/plugins/molfile/dcdplugin.html~DCD} trajectory files (Jmol 12). These files require the COORD keyword and a file giving atom information, such as a PDB file. For example: load trajectory "t.pdb" coord "t.dcd"||DGRID|{http://www.scm.com/News/DGrid.html~DGRID} file reader (Jmol 12). These files are generalized representations of output from a variety of quantum mechanical calculation packages, including especially {http://www.scm.com/Doc/Doc2009.01/ADF/ADFUsersGuide/page430.html~ADF}.||FCHK|Gaussian formatted check-point file format||FoldingXYZ|XYZ file created by the {http://fahwiki.net~Folding@home project}||GAMESS|{http://www.msg.chem.iastate.edu/gamess/~General Atomic and Molecular Electronic Structure System} output file||Gaussian|{http://www.gaussian.com/~Gaussian} output file. spin density will be read into an atoms\'s property_spin property .||GhemicalMM|{http://en.wikipedia.org/wiki/Ghemical~Ghemical} molecular mechanics file (MM1GP)||GRO|{http://www.gromacs.org~GROMACS} .gro file format ||GULP|{https://projects.ivec.org/gulp~GULP} file format||HIN|{http://www.hyper.com/~HyperChem} native file||Jaguar|National Center for Supercomputing Applications Jaguar output file ||JANA2006| Jana2006 M40/M50 files can be read, and incommensurate structures can be displayed. Commensurately and incommensureately modulated structures can be read. Not supported to date: X-harmonic and Legendre polynomial modulation specifications, crenel-orthogonalized Fourier modulation functions.||JSON| Starting with Jmol 14.0, Jmol can read (and {#.writemodel~}) {http://www.ietf.org/rfc/rfc4627.txt~JavaScript Object Notation}-formatted data. The format is compatible with that developed for {http://web.chemdoodle.com/docs/chemdoodle-json-format~ChemDoodle}, specifically implementing the "a" and "b" keys, but also adding optional keys "_is2D" and "_scale" (see below). Data must be wrapped in a JSON object named "mol": {{"mol":{{ "a":[...], "b":[...]}} }}. White space is allowed, but only after the opening seven characters, which identify the file type as JSON. [||_is2D|true if data are to be converted to 3D automatically||_scale|scale of data in x, y, and z directions. {{"x":20,"y":-20,"z":20}}, for example, indicates that the data need to be scaled down by a factor of 20, and the y axis needs to be reversed.||a|an array of atom records of the format {{"l":atomSymbol,"x":xCoord,"y":yCoord,"z":zCoord}}, where "z" and "l" (the lower-case letter "L", for "label"; defaulting to "C") are optional.||b|an array of bond records of the format {{"b":atomIndex,"e":atomIndex,"o":order}}, for "begin", "end", and optional "order" (defaults to 1). atom indexes are 0-based.||]||JCAMP-DX| Jmol can read specialized JCAMP-DX files containing ##$MODELS records and automatically sychronize user picking actions based on ##$PEAKS record information. See {misc/Jmol-JSpecView-specs.pdf~Jmol-JSpecView-specs.pdf}. ||JME|{http://www.molinspiration.com/jme/~Java Molecular Editor} file format (a 2D, not a 3D, format)||MAGRES|{http://www.ccpnc.ac.uk/pmwiki.php/CCPNC/MagresView~Magres} files ||MDTOP, MDCRD|{http://amber.scripts.edu~AMBER} Molecular dynamics topology files and associated coordinate files. ||MOL, MOL2|Accelrys (formerly Symyx, formerly Molecular Design) {http://www.mdli.com/downloads/public/ctfile/ctfile.jsp~structure data files}, including SDF and CTAB V2000 files. Starting with Jmol 14.1.12, using the forcing prefix MOL3D, as in load "MOL3D::xxx.mol" one can cause Jmol to throw an error if a 2D MOL file is attempted to be read. Starting with Jmol 14.9.2, Jmol will interpret a MOL file bond order 14 as bond order 4 (as in [Re2Cl8]2-), 15 as bond order 5 ([Mo2Cl8]4-), and 16 as bond order 6 (Mo2). ||MOLDEN|{http://www.cmbi.ru.nl/molden/molden_format.html~Molden} data file. Jmol 14.4 allows an extended version of MOLDEN files to be read that contains crystallographic information (from CASTEP), specifically [SpaceGroup], [Operators], and [Cell] or [CellAxes] (but not both). For example:
[SpaceGroup] (Number)
1
[Operators]
x, y, z
[Cell] (Angs)
2.96340697 6.51358688 -1 90.0000 90.0000 90.0000
(or)
[CellAxes] (Angs)
2.963407 0.0 0.0
0.0 6.513587 0.0
0.0 0.0 0.0
...
||MOLPRO(XML)|{http://www.molpro.net/~Molpro} structure file||Mopac|{http://openmopac.net~OpenMopac} output file (MOPOUT)||MopacGraphF|{http://openmopac.net~OpenMopac} GRAPHF output file (for molecular orbitals)||NWCHEM|Pacific Northwest National Laboratory {http://www.emsl.pnl.gov/docs/nwchem/nwchem.html~NWChem} output file||Odyssey|WaveFunction {http://www.wavefun.com/products/odyssey/odyssey.html~Odyssey} data file (ODYDATA)||Odyssey(XML)|WaveFunction {http://www.wavefun.com/products/odyssey/odyssey.html~Odyssey} XODYDATA file||PDB|{http://www.wwpdb.org/docs.html~Protein Data Bank} files. PDB reading of X-PLOR using hybrid-36 and NAMD files using hex is also supported. See {http://www.schrodinger.com/AcrobatFile.php?type=supportdocs&type2=&ident=530~} and {http://cci.lbl.gov/cctbx_sources/iotbx/pdb/hybrid_36.py~}. ||PQR|Position/Charge/Radius data file produced by the {http://apbs.sourceforge.net/~Adaptive Poisson-Boltzmann Solver} project||P2N|{http://q4md-forcefieldtools.org/Tutorial/Tutorial-1.php#3~R.E.D. input files}, also reading the AltName field and allowing label %[altName]||PSI|{http://www.psicode.org/~PSI3} output reader (coordinates only)||PyMOL PSE|{http://pymol.org~PyMOL} session files. Jmol will read most PyMOL session files, creating a very close representation of the view as seen in PyMOL 1.6. This ongoing project aims to faithfully reproduce the key features of PyMOL sessions. A representative sampling of how well this works can be found at {http://ispcbck.weizmann.ac.il/a2jmolb/browse~} and {misc/PyMOL2Jmol_browse.pdf~}. FILTER options "nosurface" and "docache" are explained below. ||QCHEM|{http://www.q-chem.com/~Q-Chem} output file||QuantumEspresso|see {http://www.quantum-espresso.org~}; XML files readable in Jmol.||SHELX|{http://shelx.uni-ac.gwdg.de/SHELX/~SHELX} output file||Spartan|WaveFunction {http://www.wavefun.com/p+I515roducts/spartan.html~Spartan} data file||SMILES|See $smilesString, above. ||SpartanSmol|WaveFunction binary {http://www.wavefun.com/products/spartan.html~Spartan} SMOL data file, including full MacSpartan Spartan directories in ZIP format||Tinker| {http://dasher.wustl.edu/tinker/~tinker} files are simple coordinate files similar to XYZ format files, but including atom type and bonding information. If the first line only contains an atom count then the filename must be preceded by tinker::.||V3000|Symyx (formerly Molecular Design) {http://www.mdli.com/downloads/public/ctfile/ctfile.jsp~V3000 Connection Table} (CTAB or SDF) data file. Starting with Jmol 14.2, Jmol supports DATA Sgroups for reading atom properties:
:> M V30 BEGIN SGROUP:> M V30 1 DAT 0 ATOMS=(1 1) FIELDNAME=pc FIELDDATA=-0.2344 :> M V30 1 DAT 0 ATOMS=(1 2) FIELDNAME=pc FIELDDATA=0.3344 :> M V30 1 DAT 0 ATOMS=(1 3) FIELDNAME=pc FIELDDATA=-0.4344 :> M V30 END SGROUP
If the field name contains "partial" then Jmol will assign atom partial charges directly. Other properties will be read as {{*}}.property_xxx, where xxx is the field name.||WebMO|{http://www.webmo.net/~WebMO} molecular orbital file reader||VASP|Vienna Ab Initio Simulation Package {http://cms.mpi.univie.a+I502c.at/vasp/~VASP} vasprun.xml, CHGCAR, and OUTCAR files. ||Wien2k|{http://www.wien2k.at~Wien2k} data files. WIEN2k performs electronic structure calculations for solids using density functional theory. Using the option spacegroup "none" disregards symmetry information given in the file and simply reads the atom coordinates given in the file, including MULT atom records. For example, load t.struct {{1 1 1}} spacegroup "none" ||XYZ|Minnesota Supercomputer Institute XMol file format. Various extensions to this file format allow reading of the following information separated by whitespace:[||element | x | y | z || element | x | y | z | vibX | vibY | vibZ || element | x | y | z | charge | | | | | (Jmol 12)|| element | x | y | z | charge | radius | | | | (Jmol 12)|| element | x | y | z | charge | vibX | vibY | vibZ | | (Jmol 12)|| element | x | y | z | charge | vibX | vibY | vibZ | atomNumber| (Jmol 12)||] If the charge is an integer, it is read as formalCharge; if it is decimal, then as partialCharge. Any information past x y z is optional, and if missing or uninterpretable as a number (for example, "X" or "--") will be ignored. The element may be either an atomic symbol or an isotope symbol such as 13C or an atomic number such as 6. ||ZMatrix|The Z-Matrix format allows for molecules to be described by internal coordinates (distances, angles, and dihedrals) rather than in terms of actual Cartesian coordinates. Jmol can read several forms of Z-matrix data, including {http://openmopac.net/manual/gaussian_z.html~MOPAC} and {http://www.gaussian.com/g_tech/g_ur/c_zmat.htm~Gaussian} format as well as a format designed for Jmol that allows also the simple introduction of bond order. Details are given in the {http://jmol.svn.sourceforge.net/viewvc/jmol/trunk/Jmol/src/org/jmol/adapter/readers/simple/ZMatrixReader.java?view=markup~Z-Matrix reader code} itself. The reader supports dummy atoms, atom labels, symbolic variable definitions, Cartesian coordinates or a mix of Cartesian and internal coordinates, alternative distance + two angles rather than distance + angle + dihedral, and specification of bond order. (Jmol 12.3.8). Gaussian and MOPAC file names must be preceded with ZMATRIX:: to bypass attempted format identification, as those file formats have no easily recognizable internal characteristics. ||]','TEXT','[File types]','') newCmd('','11.3.48','','*v+11.4;','Jmol can read specific files within compressed {#.reading ZIP/JAR files~ZIP and JAR collections}. In addition, for the load command specifically, Jmol will look for a file within the collection with the name JmolManifest and follow the directives within it. These directives may be as simple as a list of files to be loaded, one filename per line (which will be read in the order listed). Lines starting with # are comment lines, which may contain any text, but also may contain one or more of the following keywords:
[||#EXCEPT_FILES|The list of files specifies files to ignore, not files to load; all other files will be loaded.||#IGNORE_ERRORS|Try to read files, but ignore errors when a file is not recognized as a valid model format that Jmol can read. This option allows easy "mining" of ZIP collections for files that Jmol can recognize, ignoring all others. ||#IGNORE_MANIFEST|Ignore this manifest in its entirety -- simply read all files in the order retrieved by the ZIP file iterator.||]','TEXT','[ZIP/JAR files and JmolManifest]','') newCmd('','','','','The following options may be indicated after specifying the filename. Each is optional, but if more than one option is indicated, options must be given in the order listed in this table.
[||MANIFEST "manifestOptions"| If the file being loaded is a ZIP or JAR file, Jmol will search for a file in that compressed file collection with the name "JmolManifest" and process it accordingly (see above). However, in the load command, if the keyword MANIFEST and a quoted string follows the filename, then Jmol will use this string as the manifest instead, with lines of the manifest separated by vertical bar "¦" characters. In this way, standard ZIP collections can be read, and the order of file loading can be specified. For example: load "test.zip" manifest "CH3CL.MOL¦CH4.MOL" reads only these two files from the ZIP collection, in this order. If the file contains a manifest and that manifest is simply to be ignored, the quoted string should read "IGNORE_MANIFEST". || (integer) | Loads only the specified model number (if positive) or vibration number (if negative), skipping the others. For PDB files, the number indicated is the number specified in the MODEL record. (Not supported by all file types.) See also {#.load MODELS~}. ||{{unitCell(s)}} | see Crystallographic Options, below||OFFSET {{x y z}}| You can offset atoms in a file by a given amount at load time. The offset can be expressed in fractional coordinates, such as {{1/2 1/2 1/2}}, and although primarily intended for CIF file reading, can be applied to any file type. If a space group and/or unit cell are specified, the OFFSET parameter must follow that specification.||FILTER | The FILTER parameter specifies file-type specific load options (see below). Options should be separated by commas. For atom selection, more than one specification may be made using a comma between specifications: *:1,*:2, but * and ! may not be mixed within any one type (atom name, group name, or chain ID). "¦" indicates OR in a broader context: load 1blu.pdb filter "*.CA ¦ HETATM,![HOH]" selects for alpha carbons OR (HETATM and not HOH).||]','TEXT','[General Options]','') newCmd('','','','','The following crystallographic options may be indicated, starting with the specification of the unit cell. If more than one option is indicated, options must be given in the order listed in this table.
[||{{lattice}} | Jmol can read unit cell and symmetry information from selected file types (for example, CIF, PDB, or SHELX). The specific set of unit cells to load can be specified one of two ways -- either using the notation {{i j k}} or the notation {{ijk i\'j\'k\' n}}. [||{{i j k}} | Loads a block of unit cells between the origin, {{0 0 0}} and the specified unit cell system coordinate. Used alone, {{i j k}} is only for working with files containing both unit cell and space group information (CIF, SHELX, CML, for example). The particular choice {{3 3 3}} is significant, in that it loads 27 unit cells, forming a solid block around a central cell. The unit cell display can then be moved to the origin of this central cell using unitcell {{1 1 1}}, and the display of atoms can be restricted to that center cell using restrict cell=666 or restrict cell={{2 2 2}}. Multiple unit cell loading can be combined with the single-model loading by indicating the model number first, then the number of unit cells: load "myfile.cif" 15 {{3 3 3}}. Quotes are not required. There is no restriction other than memory on the size of i, j, and k (except that all must be positive). ||{{ijk i\'j\'k\' -1}} |
 Loads a block of unit cells within the range ijk and i\'j\'k\' (which should include 555) and packs all atoms into the designated set of cells. If atoms are on faces, those atoms will be duplicated at each equivalent face.||{{ijk i\'j\'k\' 0}} |
Loads a block of unit cells within the range ijk and i\'j\'k\' (which should include 555) and packs all atoms into the designated set of cells. If atoms are on faces, those atoms will be duplicated at each equivalent face.||{{ijk i\'j\'k\' 0}} |  Loads a block of unit cells within the range ijk and i\'j\'k\' (which should include 555) WITHOUT normalizing the operators. All symmetry-generated atoms are placed based on the exact definition of the symmetry operations found in the file or designated using the spacegroup keyword (see option below). Note, however, that if explicit operations are not provided and therefor must be generated from a spacegroup name, they will be normalized. The list of operations used can be obtained using {#.show~show symmetry}.||{{ijk i\'j\'k\' 1}} |
Loads a block of unit cells within the range ijk and i\'j\'k\' (which should include 555) WITHOUT normalizing the operators. All symmetry-generated atoms are placed based on the exact definition of the symmetry operations found in the file or designated using the spacegroup keyword (see option below). Note, however, that if explicit operations are not provided and therefor must be generated from a spacegroup name, they will be normalized. The list of operations used can be obtained using {#.show~show symmetry}.||{{ijk i\'j\'k\' 1}} |  Loads a block of unit cells within the range ijk and i\'j\'k\' (which should include 555), normalizing the operators to move the geometric center of the generated set of atoms into cell 555, then applying the lattice translation. Thus, load "filename" {{555 555 1}} is equivalent to load "filename" {{1 1 1}}. For example, load "myfile.cif" {{444 666 1}} loads a block of 27 unit cells, with the geometric center of all units with the bounds of the fractional coordinate range {{-1 -1 -1/}} to {{2 2 2/}}.||] ||CENTROID| The CENTROID keyword specifies that molecular cyrstal models should be created consisting of all molecules having their center of geometry, or centroid, within the specified block of unit cells (or {{1 1 1}} if not specified). The result is full molecules that are not broken across unit cell boundaries, with no duplication from one crystal face to another.||PACKED x.x| The keyword PACKED may be used in place of {{555 555 -1}} or after a designated set of cells: load t.struct {{2 2 2}} PACKED in order to load a set of atoms packed into the unit cell. If atoms are on faces, those atoms will be duplicated at each equivalent face. An optional range in fractional coordinates can be specified for including atoms that are outside the specified unit cell (default 0.02). ||SUPERCELL {{na nb nc}}| The SUPERCELL option allows reassigning the unit cell as any multiple of the actual unit cell. If this option is used, the final loaded symmetry is automatically set to P1, and cell parameters a, b, and c will be modified. A larger lattice can still be indicated, in which case they refer to how many supercells will be loaded. For example: load "quartz.cif" {{2 2 2}} supercell {{5 5 1}} will create a supercell that is 5x5x1 standard unit cells in size and then load a set of eight of those. SUPERCELL defaults to PACKED, but simply adding a lattice overrides this: load ... {1 1 1} SUPERCELL ... .||SUPERCELL "axisFormula"| This supercell option allows creating a supercell using a linear combination of the x, y, and z vectors of the file-based unit cell. If this option is used, the final loaded symmetry is automatically set to P1, and cell parameters a, b, c, alpha, beta, gamma will be modified. For example, load "quartz.cif" packed supercell "2x, x + 2y, z" will load a rectangular unit cell instead of the hexagonal cell indicated in the file. You can add an origin offset to this specification as well: load quartz.cif supercell "2a,2b+a,c;1/2,0,0" allows adjusting origin without changing symmetry operations. The supercell option is equivalent to FILTER with cell=: load quartz.cif filter "cell=2a,2b+a,c;1/2,0,0". Note that the default lattice is set to {{555 555 -1}} (i.e., PACKED) with a range based on the supercell dimensions. To indicate a specific range for packing that is not the default of 0.02, simply indicate that explicitly: load quartz.cif PACKED 0.10 supercell "2a,2b+a,c;1/2,0,0".||RANGE x.x | Restricts the atoms loaded to those within a given range in angstroms. If x.x is positive, then this range is relative to the entire set of atoms that would be generated using load "filename" {{1 1 1}}, and all atoms within the given distance from ANY atom in that set is loaded; if x.x is negative, then the range is relative to the atoms that would be generated using just load itself (the base x,y,z symmetry set), and the atoms loaded are all those within a box that is the designated distance larger than that needed to just contain the set of atoms. Then, to later select atoms actually within a given distance from the base set, use within with select or display: load 1crn.pdb {{1 1 1}} range -4; display within(4.0, symop=1555). (This two-step sequence is very efficient.) Note that Jmol will expand the load box by one number in each direction in order to find atoms that are outside of {{i j k}} but still within the designated range: load {{555 555 1}} range 2 actually checks all atoms within the 27-cell range 444 through 666. ||FILL| Fills (packs) a 10x10x10 Angstrom box (bounded by {{0 0 0}} and {{10 10 10}}) with atoms, regardless of the unit cell. This option allows comparing and visualizing crystal structures in a common range of space.||FILL x.y| fills cubic box of the specified dimension. For example, load quartz.cif FILL 20.0.||FILL BOUNDBOX| Fill the space defined by the current boundbox.||FILL UNITCELL| Fill the space defined by the current unit cell. additional options include FILL UNITCELL PRIMITIVE and FILL UNITCELL CONVENTIONAL. Usually used with load "", as the current unit cell is defined for the model currently loaded.||FILL [o vabc]| Fill a rectangular space bounded by two corners of a diagonal. For example, load quartz.cif FILL [ {{0 0 0}} {{10 10 10}} ]. Note that the parameter after FILL is an array, not just two coordinates. Fractional coordinates are allowed, and if present refer to the current unit cell, not the one being loaded. ||FILL [o va vb vc]| Fill an arbitrary unit cell defined by an origin and three crystallographic axis vectors. ||SPACEGROUP "name"| Loads a block of unit cells between the origin, {{0 0 0}} and the specified unit cell system coordinate. If the name is empty quotes, the space group of the currently loaded file will be used. In addition, the symmetry inherent in the file is ignored, and the specified space group is applied. Quotes are required around the space group name. If the space group name itself includes double quotes, use two single quotes or an "escaped double quote" (\\") instead. For example: P 32 2\'\' (single quotes here) or P 32 2\\", not P 32 2". Generally Jmol reads the Jones-Faithful operators from a file, however if the spacegroup name is specified as "ignoreOperators", Jmol will ignore any explict file-based Jones-Faithful operators and instead create the symmetry based on parsing of the space group symbol in the file (Hermann-Mauguin, Hall, or international table number). If the name is a semicolon-separated list of Jones-Faithful operators, such as "x,y,z;x+1/2,y,z", Jmol will ignore any explict file-based operators and instead create the symmetry based on the list provided. || UNITCELL [a,b,c,alpha,beta, gamma]| Specifies the unit cell to use for models in this file. If a unit cell is specified in a file, then this option allows overriding that specification. A 0 for b implies polymer (1D) periodicity (and c is ignored), otheriwse a 0 for c indicated slab (2D) periodicity. When both SPACEGROUP and UNITCELL are provided, Jmol can display molecules found in standard Cartesian coordinate files (XYZ, MOL, PDB) as packed unit cells. .|| UNITCELL [ax, ay, az, bx, by, bz, cx, cy, cz]|Specifies the unit cell in terms of the XYZ coordinates for the vectors a, b, and c. If bx = by = bz = 0, then cx, cy, and cz are ignored, and polymer (1D) periodicity is implied; otherwise, if cx = cy = cz = 0, then slab (2D) periodicity is implied. UNITCELL "" uses the unit cell of the currently loaded file. This allows, for example, adding of atoms to a crystal model using fractional coordinates:
Loads a block of unit cells within the range ijk and i\'j\'k\' (which should include 555), normalizing the operators to move the geometric center of the generated set of atoms into cell 555, then applying the lattice translation. Thus, load "filename" {{555 555 1}} is equivalent to load "filename" {{1 1 1}}. For example, load "myfile.cif" {{444 666 1}} loads a block of 27 unit cells, with the geometric center of all units with the bounds of the fractional coordinate range {{-1 -1 -1/}} to {{2 2 2/}}.||] ||CENTROID| The CENTROID keyword specifies that molecular cyrstal models should be created consisting of all molecules having their center of geometry, or centroid, within the specified block of unit cells (or {{1 1 1}} if not specified). The result is full molecules that are not broken across unit cell boundaries, with no duplication from one crystal face to another.||PACKED x.x| The keyword PACKED may be used in place of {{555 555 -1}} or after a designated set of cells: load t.struct {{2 2 2}} PACKED in order to load a set of atoms packed into the unit cell. If atoms are on faces, those atoms will be duplicated at each equivalent face. An optional range in fractional coordinates can be specified for including atoms that are outside the specified unit cell (default 0.02). ||SUPERCELL {{na nb nc}}| The SUPERCELL option allows reassigning the unit cell as any multiple of the actual unit cell. If this option is used, the final loaded symmetry is automatically set to P1, and cell parameters a, b, and c will be modified. A larger lattice can still be indicated, in which case they refer to how many supercells will be loaded. For example: load "quartz.cif" {{2 2 2}} supercell {{5 5 1}} will create a supercell that is 5x5x1 standard unit cells in size and then load a set of eight of those. SUPERCELL defaults to PACKED, but simply adding a lattice overrides this: load ... {1 1 1} SUPERCELL ... .||SUPERCELL "axisFormula"| This supercell option allows creating a supercell using a linear combination of the x, y, and z vectors of the file-based unit cell. If this option is used, the final loaded symmetry is automatically set to P1, and cell parameters a, b, c, alpha, beta, gamma will be modified. For example, load "quartz.cif" packed supercell "2x, x + 2y, z" will load a rectangular unit cell instead of the hexagonal cell indicated in the file. You can add an origin offset to this specification as well: load quartz.cif supercell "2a,2b+a,c;1/2,0,0" allows adjusting origin without changing symmetry operations. The supercell option is equivalent to FILTER with cell=: load quartz.cif filter "cell=2a,2b+a,c;1/2,0,0". Note that the default lattice is set to {{555 555 -1}} (i.e., PACKED) with a range based on the supercell dimensions. To indicate a specific range for packing that is not the default of 0.02, simply indicate that explicitly: load quartz.cif PACKED 0.10 supercell "2a,2b+a,c;1/2,0,0".||RANGE x.x | Restricts the atoms loaded to those within a given range in angstroms. If x.x is positive, then this range is relative to the entire set of atoms that would be generated using load "filename" {{1 1 1}}, and all atoms within the given distance from ANY atom in that set is loaded; if x.x is negative, then the range is relative to the atoms that would be generated using just load itself (the base x,y,z symmetry set), and the atoms loaded are all those within a box that is the designated distance larger than that needed to just contain the set of atoms. Then, to later select atoms actually within a given distance from the base set, use within with select or display: load 1crn.pdb {{1 1 1}} range -4; display within(4.0, symop=1555). (This two-step sequence is very efficient.) Note that Jmol will expand the load box by one number in each direction in order to find atoms that are outside of {{i j k}} but still within the designated range: load {{555 555 1}} range 2 actually checks all atoms within the 27-cell range 444 through 666. ||FILL| Fills (packs) a 10x10x10 Angstrom box (bounded by {{0 0 0}} and {{10 10 10}}) with atoms, regardless of the unit cell. This option allows comparing and visualizing crystal structures in a common range of space.||FILL x.y| fills cubic box of the specified dimension. For example, load quartz.cif FILL 20.0.||FILL BOUNDBOX| Fill the space defined by the current boundbox.||FILL UNITCELL| Fill the space defined by the current unit cell. additional options include FILL UNITCELL PRIMITIVE and FILL UNITCELL CONVENTIONAL. Usually used with load "", as the current unit cell is defined for the model currently loaded.||FILL [o vabc]| Fill a rectangular space bounded by two corners of a diagonal. For example, load quartz.cif FILL [ {{0 0 0}} {{10 10 10}} ]. Note that the parameter after FILL is an array, not just two coordinates. Fractional coordinates are allowed, and if present refer to the current unit cell, not the one being loaded. ||FILL [o va vb vc]| Fill an arbitrary unit cell defined by an origin and three crystallographic axis vectors. ||SPACEGROUP "name"| Loads a block of unit cells between the origin, {{0 0 0}} and the specified unit cell system coordinate. If the name is empty quotes, the space group of the currently loaded file will be used. In addition, the symmetry inherent in the file is ignored, and the specified space group is applied. Quotes are required around the space group name. If the space group name itself includes double quotes, use two single quotes or an "escaped double quote" (\\") instead. For example: P 32 2\'\' (single quotes here) or P 32 2\\", not P 32 2". Generally Jmol reads the Jones-Faithful operators from a file, however if the spacegroup name is specified as "ignoreOperators", Jmol will ignore any explict file-based Jones-Faithful operators and instead create the symmetry based on parsing of the space group symbol in the file (Hermann-Mauguin, Hall, or international table number). If the name is a semicolon-separated list of Jones-Faithful operators, such as "x,y,z;x+1/2,y,z", Jmol will ignore any explict file-based operators and instead create the symmetry based on the list provided. || UNITCELL [a,b,c,alpha,beta, gamma]| Specifies the unit cell to use for models in this file. If a unit cell is specified in a file, then this option allows overriding that specification. A 0 for b implies polymer (1D) periodicity (and c is ignored), otheriwse a 0 for c indicated slab (2D) periodicity. When both SPACEGROUP and UNITCELL are provided, Jmol can display molecules found in standard Cartesian coordinate files (XYZ, MOL, PDB) as packed unit cells. .|| UNITCELL [ax, ay, az, bx, by, bz, cx, cy, cz]|Specifies the unit cell in terms of the XYZ coordinates for the vectors a, b, and c. If bx = by = bz = 0, then cx, cy, and cz are ignored, and polymer (1D) periodicity is implied; otherwise, if cx = cy = cz = 0, then slab (2D) periodicity is implied. UNITCELL "" uses the unit cell of the currently loaded file. This allows, for example, adding of atoms to a crystal model using fractional coordinates:set appendNew false
LOAD DATA "append"
1
testing
Na 0.5 0 0.5
end "append" {{1 1 1}} spacegroup "" unitcell ""
||UNITCELL "a=...,b=...,c=...,alpha=...,beta=...,gamma=...."| Specifies the unit cell from a string such as "a=10,b=10,c=20,alpha=90,beta=90,gamma=129".||]','TEXT','[Crystallographic Options]','') newCmd('','','','','The load command by itself reloads the current file.','*10','','') newCmd('','','','','Jmol automatically determines file type based upon the contents of the file. Quotes are recommended. Files containing fractional coordinates are displayed with their unit cell visible. load "" reloads the current file. For files containing multiple models, an optional integer after the file name will load only the specified model. If this number is negative, it refers to a specific vibrational mode in files that contain that information . If this number is 0, it refers to the last model in the set . In the case of PDB and mmCIF files, this number refers to the model number specied in the MODEL record or _atom_site.pdbx_PDB_model_num field, respectively.','*20','"filename"','(integer)') newCmd('','','','*v+12.2;','An additional way to load a specific set of models involes using an array just after the file name, starting with model 1 for the first. The order of models is not significant. Thus, [1 2 3] is the same as [3 2 1] and loads the first three models only. (Note that if you want to display models in a different sequence then they appear in a file, you can do that with the {#.frame~} command.) In the case of PDB and mmCIF files, these numbers refer to the model number specied in the MODEL record or _atom_site.pdbx_PDB_model_num field, respectively. A comparison of load options using just a model number or an array is given below:[||load "" |loads the first model in a file when not PDB or mmCIF or the model with MODEL 1 record for a PDB file or the model with _atom_site.pdbx_PDB_model_num = 1 for an mmCIF file.||load "" [1] |same as load "" 1; brackets allow for more than one model.||load MODELS ({{1}}) ""|(see {#.loadmodels~load MODELS}) always loads the SECOND model in a file, regardless of its indicated model number.||]','*21','"filename"','[i j k l m...]') newCmd('','','','','File format can be forced by prefixing a filename with "xxxx::" where "xxxx" is a Jmol file type. This should not be necessary in most cases, since Jmol determines file type by scanning the first lines of a file for file-type-specific content. In certain cases, however, where there are extensive comments at the beginning of a file, or a file type (such as MDCRD) does not have any distinguishing characteristics, it is possible for Jmol to fail to discover the file type or to misassign it. In that case, adding "xxxx::" is necessary. ','*31','"filetype::filename"','') newCmd('','','','','Loads the file with the name specified by variable variableName. You can load a set of files that are defined in an array variable.','*33','@variableName','') newCmd('','12.0.?','','*v+12.0;','Loads the file data given in quotes. Generally this would be a small molecule in JME or XYZ format.','*40','INLINE','"fileData"') newCmd('','','','','You can load a model from data contained in a variable. This allows modification of the data prior to display, for example. Quotes are necessary, as without them -- load @x -- it is the file name that is expected in variable x, not the file contents. For example: x = load("quartz.cif");load "@x" {{2 2 2}};reset x. Note that to save memory, it is a good idea to clear the variable using reset x just after loading. An interesting aspect of this option is that it allows data from a remote file to be saved in the state. This means that if you use write state "somefile.spt", then that single state script will contain the full file data in a DATA statement. It will be "transportable" and no additional model file will be required. (See also the PNGJ format for the {#write~} command, which is even better.)','*41','"@variableName"','') newCmd('','14.3.16','','*v+14.4;','Loads the file data given in variable x; same as load "@x" but in the same syntax as {#.write~write VAR x}.','*42','VAR','x') newCmd('','14.2.6','','*v+14.4;','You can load mmCIF files directly from {http://www.ebi.ac.uk/pdbe~}. Simply preface the four-letter PDB id code with "*", and Jmol will load the file. As for other file laoding, you can adding "AS ." ("as" with a period) you can save that file automatically in the default directory (as xxxx.cif), and using, for example, load *1crn AS "myfile.cif", you can save it to some other local file name. Starting with Jmol 14.4, you can add "/dssr" to the PDB id to also read RNA/DNA secondary structure information from Columbia University, "/rna3d" for RNA secondary structure annotations from {http://rna.bgsu.edu/rna3dhub~}, {#.domainssequencedomainannotations~sequence domain annotations} using "/dom", and {#.domainsstructurevalidationannotations~structure validation annotations} using "/val".','*4310','*XXXX','') newCmd('','14.3.16','','*v+14.4;','Appending a * to a PDBe ID loads a PDBe "updated" CIF file, which allows PDB CONECT-like bond creation. An example of such a file can be found {http://www.ebi.ac.uk/pdbe/static/entry/1h68_updated.cif~ here}. This is a suitable replacement for PDB CONECT, integrating information found in {http://mmcif.wwpdb.org/dictionaries/mmcif_mdb.dic/Items/~_chem_comp_bond} and {http://mmcif.wwpdb.org/dictionaries/mmcif_mdb.dic/Items/_struct_conn.conn_type_id.html~_struct_conn} categories. Note that the presence of _chem_comp_bond records in these files will enable processing of _struct_conn as well, regardless of filter "addbonds".','*4311','*XXXX*','') newCmd('','11.1.22','','','You can load PDB files directly from {http://www.rcsb.org~} or another server of your choice. (This depends upon the setting of {#.set(files and scripts)~set loadFormat}). Simply preface the four-letter PDB id code with "=", and Jmol will load the file. Adding "AS ." ("as" with a period) you can save that file automatically in the default directory (as xxxx.pdb.gz), and using, for example, load =1crn AS "myfile.pdb", you can save it to some other local file name. The default is to transfer the file in g-zipped format; If you add ".pdb" to the expression -- load =1crn.pdb, for example -- then Jmol will transfer and save the uncompressed PDB file. Starting with Jmol 14.2, you can add "/dssr" to the PDB id to also read RNA/DNA secondary structure information from Columbia University. Starting with Jmol 14.4, you can load "/rna3d" for RNA secondary structure annotations from {http://rna.bgsu.edu/rna3dhub~}, {#.domainssequencedomainannotations~sequence domain annotations} using "/dom", and {#.domainsstructurevalidationannotations~structure validation annotations} using "/val".','*432','=XXXX','') newCmd('','11.1.22','','','You can load PDB ligand (chemical component) files directly from {http://www.rcsb.org~} or another server of your choice. (This depends upon the setting of {#.set(files and scripts)~set loadLigandFormat}). Simply preface the three-letter PDB id code with "==", and Jmol will load the file. ','*441','==XXX','') newCmd('','13.1.13','','','You can load files from a named database. Options (Jmol.4.6) include:[|| mp |load =mp/24972| loads a file from {http://www.materialsproject.org~}|| ligand | load =ligand/hem | RCSB chemical component file|| nci | load =nci/CC | NCI CACTVS Resolver|| pdb | load =pdb/1crn | RCSB pdb files|| iucr| load =iucr/wf5113sup1 | IUCr Acta Cryst B supplemental files|| cod| load =cod/1000002 | {http://www.crystallography.net/cod~Crystallography Open Database}|| iucr| load =pdbe/1crn | CIF file from {http://www.ebi.ac.uk/pdbe~PDBe}|| iucr| load =pdbe2/1crn | CIF file from {http://www.ebi.ac.uk/pdbe~PDBe} (alternative)|| aflow| load =aflow/AgAu | {http://aflowlib.org~AFLOW} binary metal alloy file|| magndata| load =magndata/1.1.3 |{http://webbdcrista1.ehu.es/magndata~MAGNDATA} magnetic crystal database (Bilbao Crystallographic Server|| ams| load =ams/quartz | {http://rruff.geo.arizona.edu/AMS~American Mineralogist} Crystal Structure Database||]','*442','=database/id','') newCmd('','12.3.23','','','You can load models from PubChem. Preface the compound name with colon. Quotes are only required if nonalphanumeric characters are present in the name.','*443','":chemical name"','') newCmd('','12.0.RC15','','','You can load SMILES strings, and Jmol will turn them into 3D models using the {https://cactus.nci.nih.gov~NIH Cactus server}. As for reading files from any source outside your domain, you will have to use the signed applet or Jmol application to do this. These files can be saved as MOL files using {#.write~write xxx.mol} or load $xxxx AS "myfile.mol", and if the conformation is not to your liking, switching to {#.set modelkitMode~} or using {#.set picking~set picking dragMinimize} you can quickly adjust the model to the desired conformation. Quotation marks should be used for names that include the space character: load "$ethyl acetate".','*45','$smilesString','') newCmd('','12.0.RC15','','','The service at {https://cactus.nci.nih.gov~} is not limited to SMILES strings. The database includes several million known compounds, which are accessible by CAS registry number, InChI codes, and chemical name as well. For example, load $57-88-5 loads a model of chlolesterol, and load $taxol as "taxol.mol" loads a model of taxol and saves it on your hard drive. This service taps into the {http://opsin.ch.cam.ac.uk~OPSIN} service of Cambridge University to return structures from IUPAC or IUPAC-like names. ','*460','$identifier','') newCmd('','12.0.RC15','','','Using just LOAD $ will load the current structure from {https://cactus.nci.nih.gov~NCI/CADD} by passing a SMILES string to that service. The NCI/CADD service will create 3D models directly from SMILES using {https://www.mn-am.com/products/corina~CORINA}, if necessary.','*461','$','') newCmd('','12.0.RC15','','','Using just LOAD : will load the current structure from PubChem by passing a SMILES string to that service. Note that PubChem, unlike NCI/CADD, will only deliver models that have been deposited in its database. ','*462',':','') newCmd('','14.1.16','','','The ORIENTATION keyword specifies that after the file is loaded, the orientation should be returned to its original value. This orientation can be restored later using restore orientation PRELOAD.','*47','ORIENTATION','') newCmd('','12.0.RC15','','','An alternative to the $ syntax for loading SMILES strings.','*48','SMILES','"smilesString"') newCmd('','','','','An optional keyword, {#.loadappend~APPEND}, {#.loaddata~DATA}, {#.loadfiles~FILES}, {#.loadmenu~MENU}, {#.loadmodels~MODELS}, or {#.loadtrajectory~TRAJECTORY}, may be supplied prior to the quoted filename. Other keywords are ignored. (Jmol does not use the Chime-style keyword to specify "file format". Rather, file format can be forced by prefixing a filename with "xxxx::" where "xxxx" is a Jmol file type. However, this should not be necessary in most cases, since Jmol determines file type by scanning the first lines of a file for file-type-specific content. (In certain cases, where there are extensive comments at the beginning of a file, it is possible for Jmol to fail to discover the file type or to misassign it. In that case, xxxx:: should be used.)','*11','keyword','"filename"') newCmd('','14.3.14','','*v+14.4;','The FILL option, described more fully under {#.loadcrystallographicoptions~Crystallographic Options}, allows filling an arbitrary space with a set of atoms from a crystallographic file. When the file has no unit cell, this option simply loads the file with the specified boundbox.','*60','"filename"','FILL','...','') newCmd('','','','*v+11.6;','For individual file types, it is possible to filter the data in the file as it is loaded. The FILTER keyword followed by a quoted string allows for this. The FILTER keyword must be the last keyword in the LOAD command. Specific filters include: [||file type | filter |description|| MOL | FILTER "2D"| indicates to consider the file a 2D file and to apply a automatic hydrogen addition and 2D-to-3D conversion immediately upon loading. "2D-noMIN" does the hydrogen addition but no minimization. ||CIF, GROMACS, MDTOP, PDB, PQR | FILTER "[XXX], .XXX, :X, %X"| to specify inclusion or exclusion of specific residue types, atom types, chains, or alternative locations. The prefix "!" indicates NOT. Within each of these four sets, multiple selections are treated as "OR" without "!" and "AND" when "!" is present. With % (alternative location), atoms with no alternative location marking are ALWAYS loaded regardless of this filter. For example, load "1sva.pdb" FILTER "*.CA,%B" loads only alpha carbons that correspond to the conformation involving alternative location B; FILTER "![HOH]" filters out water molecules.|| GAMESS, GAUSSIAN, GenNBO, Jaguar, Molden, NWChem, PSI, and QCHEM | FILTER "xxx"| where "xxx" is a word on the line Jmol uses to identify a molecular orbital. This allows selective loading of specific types of molecular orbitals -- such as "alpha", "beta", or "NBO" -- for any of these file types, "POPULATION" or "EDMISTON" or "PIPEK" for GAMESS. ("NBO" refers to orbitals generated with the NBO 5.0 option AONBO=P; "EIGEN" or "!NBO" will skip loading of these orbitals.)||AFLOW | FILTER CA=x.x Select from the binary alloy file for alloy AaBb only models with the specified fractional concentration of Aa.||AFLOW | FILTER CB=x.x Select from the binary alloy file for alloy AaBb only models with the specified fractional concentration of Bb.|| crystallographic files |
load "myfile.pdb";load APPEND "myligand.xyz";frame *;display 1.1,2.1
If set appendNew false has been issued and only one model is in the appended file, then the file model is added to the currently displayed model, creating no additional frames. Multiple files may be appended if the specified file is a ZIP file. In that case, specifying the manifest in the command after the filename is an option. ','1|2','','') newCmd('.load DATA','12.0.RC24','','*v+12.0;','The DATA "model..." and DATA "append..." commands are deprecated in favor of load DATA.... All of the parameter options for the load command are thus also available for the {#.data~} command. ','1|2','','') newCmd('.load [property]','','','*v+11.8;vib','Adding the keyword OCCUPANCY, PARTIALCHARGE, TEMPERATURE, VIBRATION, or XYZ to the load command instructs Jmol to load only that sort of information from a file.
[||load OCCUPANCY| Occupancy data are in the form of integers between 0 and 255. Numbers less than 0 are saved as 0; numbers greater than 255 are saved as 255.||load PARTIALCHARGE| Partial charge data are in the form of decimals roughly in the range +/-3.4 x 1038.||load TEMPERATURE| Temperature (B-Factor) data are saved as decimals with 0.01 precision within the range -327.68 to 327.67. || load VIBRATION | Vibrational data are in the form of vectors. ||load XYZ | Loads only the coordinates, replacing the current coordinates of an already-loaded model with those in the file to be read. ||]
The data are applied to the currently selected set of atoms based solely on atom position. All standard load parameters are accepted, although many will be ignored, however whereas the default for a normal LOAD operation is to load all files, the default is to read data only from the first model in a multi-model file (or the specific model indicated with an integer after the file name). For each "atom" position and vector that is read, Jmol applies the data to all selected atoms having a unit cell normalized position within {#.set (misc)~loadAtomDataTolerance} (default 0.01) Angstroms of the position read from the file. If the file being loaded contains embedded Jmol script commands, those commands will be processed after the application of the data. For example, load "myfile.struct" {{5 5 1}} PACKED; select _O; set loadAtomDataTolerance -0.2; load VIBRATION "vibs.xyz" 3 first loads a set of unit cells from myfile.struct, then applies only to the oxygen atoms the third vibration set found in vibs.xyz. Oxygen atoms in all unit cells will be given data even though the data in vibs.xyz might only be for one unit cell. ','1|2','','') newCmd('.load AUDIO','','','','Jmol can load and play audio files of standard types. JavaScript: WAV, MP3, OGG; Java: WAV','1|2','','') newCmd('','','','','Load and play an audio file.','*1','"filename"','','....','') newCmd('.load FILES','','','*v+11.2;','Adding FILES to the load command indicates that a set of files are to be loaded . Filenames must be quoted. Each model encountered is put into a new "frame." No additional parameters other than COORD are allowed (see below). ','1|2','','') newCmd('','','','','For multiple file loading, all parameters after the first must appear in quotes. Each model is loaded into a new frame, and frames are addressed using a decimal notation -- 1.1 for the first model in the first file, 1.2 for the second model in the first file, 2.1 for the first model in the second file, etc. For example, select (oxygen) and (1.3, 1.4) selects all oxygens in the third or fourth model of the first file loaded. See also {#.set(misc)~set backgroundModel}. The special command load MENU allows loading of a custom Jmol popup menu. ','*1','"filename1"','"filename2"','....','') newCmd('','14.1.13','','*v+14.2;','If a + is used between file names, Jmol will load the files as a single stream based on the first file\'s type. This can be useful when part of the required information for the file loading is in a second file. For example, if a PDB file and its associated DSSR data are in two files, one can use load files "xxx.pdb"+"xxx.dssr" to load the structure and process the DSSR data. (One can also use load =1crn/dssr do accomplish this particular task if the PDB and DSSR files are not available locally.)','*2','"filename1"','+','"filename2"','') newCmd('.load HISTORY','','','*v+11.4;','The special command load HISTORY allows loading of the command history from a file. ','1|2','','') newCmd('','','','','The history file can contain any Jmol script.','*1','"filename"','') newCmd('.load MENU','','','*v+11.4;','The special command load MENU allows loading of a custom Jmol popup menu. ','1|2','','') newCmd('','','','','File format is that written by Jmol using the {#.show~show MENU} or {#.write~write MENU} command. See examples at {misc/Jmol.mnu~misc/Jmol.mnu} and {misc/test.mnu~misc/test.mnu}. Set the label for a menu to null to remove a menu item or submenu.','*1','"menufile"','') newCmd('.load MODELS','','','*v+11.8;','The MODELS keyword allows loading of a specific subset of models from a file that contains a model collection. Two syntaxes are available. Note that model numbers refer to the sequence of models encountered in the file, starting with 0, and typically do NOT correspond to the model numbers indicated in PDB MODEL records. Once the models are loaded, they can be accessed using the file.model decimal notation as 1.1, 1.2, 1.3, etc.
An additional way to load selected models is to put the desired model numbers in an array that is given AFTER the file name. In this case, this first model is "1", not "0", and if the file is a PDB or mmCIF file, the numbers refer to the model numbers specified in the file, which might not be sequential.','1|2','','') newCmd('','','','','In the first syntax, first and last models along with a "stride" (step) are specified. Models are then read using an equivalent of for (i = first; i <= last; i = i + stride). Numbers start with 0 for the first model; -1 for last indicates "to the end of the file." For example, load MODELS {{0 10 2}} "..." would load six models, models 0, 2, 4, 6, 8, and 10.','*1','{{first last stride}}','"filename"') newCmd('','','','','In the second syntax, a list of the specified files to load is given in the Jmol "bitset" syntax. This syntax uses braces within parentheses. Specific models are listed, starting with 0 for the first model. Ranges of models are indicated using colons. For example, load MODELS ({{0 2 4:6}}) "..." loads five models -- models 0, 2, 4, 5, and 6. load MODELS ({{1}}) "..." loads the second model only.','*2','({{i j k:l m...}})','"filename"') newCmd('.load TRAJECTORY','','','*v+11.4;','Loads a file as a "trajectory", meaning a single model with a series of atom coordinates. (Similar to the sort of multiple-frame animations that the Chime plug-in was able to load.) Loading a file as a trajectory saves substantially on memory requirements, since there is only one set of atoms and bonds. The defining restriction for trajectories is that only one frame can be viewed at a time. Each trajectory is loaded into its own frame as though it were a distinct model, and frames are accessed as usual using the {#.frame~} or {#.model~} command. In addition, any reference to a specific trajectory, such as select 1.3, switches to that set of coordinates, and {#.measure~Measurements} and the position of {#.cartoon~cartoons} are automatically recalculated when a new trajectory is displayed. Changes to colors in one trajectory effect the same change for all trajectories, since there is really only one model, just different atom positions. Multiple files may be loaded as independent trajectories using APPEND TRAJECTORY in place of simply TRAJECTORY. Jmol allows reading of PDB files that contain no MODEL line and are instead simply concatentated versions of the same atoms as trajectories using the TRAJECTORY keyword. Each model must start with atom number 1 for this to work. The TRAJECTORY keyword also applies to vibrations.','1|2','','') newCmd('','','','*v+11.4;','Loads the specified file as a trajectory.','*1','"filename"','') newCmd('','','','*v+11.8;','Loads the specified subset of models from the file as a trajectory. See {#.load MODELS~} for details.','*2','{{first last stride}} or ({{i j k:l m...}})','"filename"') newCmd('','','','*v+11.8;','The presence of a COORD keyword indicates that the trajectory is to be built from a set of files that includes an AMBER molecular dynamics topology file and associated coordinate files. FILTER is optional, but recommended. For example, FILTER "![WAT]" prevents loading of water molecules. Any number of coordinate files may be specified. Coordinates are loaded as a set of trajectory steps. At least one COORD keyword must be given, and each specification of first, last, and stride must be preceded by a COORD keyword. {{first last stride}} or ({{i j k:l m...}}) is optional, and if not provided defaults to {{0 -1 1}}, which is interpreted as "all trajectory steps from each file." See {#.load MODELS~} for documentation on {{first last stride}} and ({{i j k:l m...}}).','*3','"filename"','FILTER','"filter specification"','COORD {{first last stride}} or ({{i j k:l m...}}) mdcrd::crdfile1') newCmd('.log','11.9.24','','*v+12.0 -- new;','Logs data to a file. Specifically for the signed applet and the application. The LOG command works the same as {#.print~} but records the information in a log file. If the string includes "$NOW$", those characters are replaced with the current date and tims. For example: log "$NOW$ " + _version. The string "$CLEAR$" clears the log file. The file to log to must first be designated using set logFile "someName". This name will be prepended with "JmolLog_" and must not contain any directory path. The file will always be created in the Jar file directory. Note that logging is not possible with the web-based version, even with the signed applet, but signed applet or application running locally can log to a file. In addition to explicit use of the LOG command, two settings, set logCommands and set logGestures allow automatic tracking of commands and gestures (swipe, pinch, zoom, spin) to the designated log file.','1|2','','') newCmd('.loop','','','*v+11.0 -- changes how scripts are interrupted;pause','Causes the script to restart at the beginning, with an optional time delay. See also {#.goto~}.','1','.on','') newCmd('','','','','','1','._time_delay','') newCmd('.mapProperty','','','*v+12.0 -- new;','The mapProperty command allow copying of an atomic property from one set of atoms to another. The operation involves identifying two sets of atoms and associated properties and also a common "key" property such as atomno or resno. If no key is given, atomno is assumed. A shortcut allows quick transfer of atom selection.','0|1','','') newCmd('','','','','For each atom Y in atomExpression2 that matches an atom X in atomExpression1 based on propertyKey, Y.property2 is made to equal X.property1. Property2 must be settable. For example, mapProperty {{1.1}}.temperature {{2.1}}.property_t atomno; color {{2.1}} property_t "rwb" , which would color atoms in model 2.1 (perhaps a {#.plot~}) based on temperature values for model 1.1. ','*1','{{atomExpression1}}.property1','{{atomExpression2}}.property2','propertyKey','') newCmd('','','','','This form of the mapProperty command is a shortcut for mapProperty {{selected}}.selected {{atomExpression}}.selected propertyKey. For example, mapProperty SELECTED {{2.1}} selects atoms in model 2.1 that match atomno with atoms that are already selected.','*2','SELECTED','{{atomExpression}}','propertyKey','') newCmd('.measure','','','*v+11.0 -- adds several new capabilities;','Renders a measurement between the specified atoms. See also {#.set (measure)~}. Two general syntaxes are available. In the older syntax, a series of two to four atom numbers are given, and the appropriate measure (distance, angle, or dihedral angle) is then displayed. The newer, more general syntax is as follows:
measure RANGE <minValue> <maxValue> ALL|ALLCONNECTED|DELETE (<atom expression>) (<atom expression>) ...
Using this syntax one can specify a set of measurements to define all at once. Note that these sets are embedded {#.atom expressions~atom expressions} that must be enclosed in parentheses. If neither ALL nor ALLCONNECTED is present, only the first matching atom in the entire model set (all frames, so probably the first frame) is matched in each atom expression. When ALL or ALLCONNECTED is specified, all matching criteria in all frames are generated, thus allowing for "animated" measures. In general, this syntax restricts measurements to within the same model. However, measures can also be between two atoms in different frames (different models) as long as each atom expression evaluates to a single, specific atom. (To specify a particular atom in a particular model, use "AND */n", where n is the model number, "ATOMNO=3" by itself, for example, will indicate the third atom in each model/frame, but "ATOMNO=3 and */6" specifies only atom 3 in model 6). If a measurement is made between atoms in different models, both models must be displayed in order for the measurement to appear. A simple way to display two specific models is to use {#.display~display */i or */j}, where i and j are two model numbers.
Thus, for example, measure (*) (*) will measure nothing, because both expressions will simply match the first atom in the first frame. measure allconnected (*/3) (*/3) (*/3) will measure every angle associated with bonds for model 3; measure allconnected (*) (*) will measure every bonded distance in every loaded model; measure all (*) (*) measures all possible interatomic distances in all models (not recommended!).
For the applet, using {#.getProperty~getProperty measurementInfo} will then deliver full information relating to all measurements.
The format of the label that accompanies the measurement line can be set for previously created measurements using measure "n:labelFormat", described below. In addition, the default format (affecting only future-created labels) for measurement labels can be set using {#.set_defaultdistancelabel~set defaultDistanceLabel}, {#.set_defaultanglelabel~set defaultAngleLabel}, and {#.set_defaulttorsionlabel~set defaultTorsionLabel}. The {#.font~} of measurements can be set, for example, using {#.font~font measure 20}.
You can specify units for a specific label using "//" at the end of the format followed by a unit designation such as A or nm. Special units are available for NMR data arising from Magres files: "2://dc_hz" or "2://dc_khz" or "2://khz" for calculated dipolar constant in Hz or kHz and "2://isc_hz" or "2://hz" for calculated J-coupling constants. These units are also available for the measure() function as, for example, x = measure({{a}},{{b}},"isc_hz").','0|1|2|3|4','','') newCmd('','','','','Turns on and off the distance, angle, dihedral measurement labels and measurement lines. (To turn off just the labels, use {#.set measurement~set measurement OFF} ','*11','._on_off{"ON"}','') newCmd('','','','*v+11.0;','Changes all previously defined measurement labels of a given type (n = 2, 3, or 4) to the indicated format. The default label is "%VALUE %UNITS" for all types. Specifically for distances, the suffix //A will use angstroms for the default label for distance measurements. Other suffixes include //nm, //pm, and //vdw. Also available is %#(percent number-sign), which gives the 1-based number of the measurement. Atom information can be included as for {#.label~labels}, adding 1 or 2 to the format code to indicate which atom. So, for example, measure "2:%a1 -- %a2 distance = %0.0VALUE" delivers the two atom names along with the value of the measurement rounded to the nearest integer with no units indicated. Specific units for the measurements can be given after two slashes: measure "2:%a1 -- %a2 distance = %0.1VALUE //A" would display the distance rounded to the nearest 0.1 Angstrom. ','*14','"n:labelFormat"','') newCmd('','','','','Two atoms specify a distance measurement with an optionally given format. This syntax selects the first atom in the current {#.frame~} set.','*2','(integer)','(integer)','"labelFormat"','') newCmd('','','','','Three atoms specify an angle measurement. The format is optional. This syntax selects the first atom in the current {#.frame~} set.','*3','(integer)','(integer)','(integer)','"labelFormat"') newCmd('','','','','Four atoms specify a dihedral angle measurement. The format is optional. This syntax selects the first atom in the current {#.frame~} set.','*4','(integer)','(integer)','(integer)','(integer) "labelFormat"') newCmd('','','','*v+11.0;','Show the distance, angle, or dihedral angle formed by the FIRST atom in each atom expression. The format is optional. ','*15','(two to four atom expressions, each in parentheses)','"labelFormat"') newCmd('','','','*v+11.0;','Show the distance, angle, or dihedral angle formed by ALL atoms in the first expression with ALL atoms of each additional atom expression. The format is optional. ','*6','ALL','(two to four atom expressions)','"labelFormat"','') newCmd('','','','*v+11.0;','Show the distance, angle, or dihedral angle formed by ALL atoms in the first expression with ALL atoms of each additional atom expression, provided they form a connected set. The format is optional. ','*7','ALLCONNECTED','(two to four atom expressions)','"labelFormat"','') newCmd('','','','*v+11.0;','Deletes all measurements.','*81','DELETE','') newCmd('','','','*v+11.0;','Deletes a specific measurement, in order of their creation, starting with 1.','*82','DELETE','(integer)') newCmd('','','','*v+11.0;','Deletes all matching distance, angle, or dihedral angle measurements that are currently defined based on the atom expressions.','*83','DELETE','(two to four atom expressions)') newCmd('','12.1.26','','*v+12.2;','Lists all measurements. (Same as {#.show~show measurements}.)','*84','LIST','') newCmd('','','','*v+11.0;','Adding RANGE and two decimal numbers modifies the above commands to limit the measurements created or deleted to only those within this specific range of values in Angstroms (distance) or degrees (angles). The word "RANGE" itself is optional but recommended.','*90','RANGE','(decimal)','(decimal)','ALL|ALLCONNECTED|DELETE (two to four atom expressions, each in parentheses)') newCmd('','','','*v+11.0;','The SEARCH (or SMARTS) option for MEASURE allows powerful atom selection using {https://jcheminf.biomedcentral.com/articles/10.1186/s13321-016-0160-4~Jmol SMARTS}, particularly involving braces {{ }} to designate the desired two, three, or four atoms. For example, set measurementUnits HZ; measure SEARCH "{{[H]}}CC{{[H]}}" will, in Jmol 14.30.0, display all three-bond coupling constants calculated for a compound, because {{ }} indicate to do the full match but only return these specific atoms from that search. ','*91','SEARCH','"SMARTstring"') newCmd('','11.9.12','','*v+12.0;ticks','Creates a measure line with ticks along it. There are three levels of ticks - major, minor, and "subminor." Only the major ticks have labels. Which of these tick levels are displayed and the distance between ticks depends upon the parameter that takes the form of a point. The optional keyword FORMAT allows formating of the labels for the major ticks. These are based on an array of strings given after the FORMAT keyword. If the array is shorter than the number of ticks, the formats in the array are repeated. Following that, the optional keyword SCALE allows setting the scale either for each axis direction independently {{scaleX, scaleY, scaleZ}} or overall (as a decimal number). An optional keyword FIRST allows setting of the initial value of the measure. Finally, two points must be indicated. ','*93','TICKS X|Y|Z','{{major,minor,subminor}} FORMAT ["%0.2f", ...] ','SCALE {{scaleX, scaleY, scaleZ}} | x.xx','FIRST x.xx {{point1}} {{point2}}') newCmd('.measure ID','','','*v+13.0;','Using a measurement ID option allows you more control over how the measurement renders as well as the ability to turn it on and off selectively. As for MEASURE without ID, you can specify from two to four atoms or points to be the basis for the measurement. If text is not given, then the numerical value of the measurement is used and the id is for a measurement group. Thus, more than one measurement without text may have the same ID. IDs without spaces do not require quotes, but since the parser will sometimes subsititute known keywords ("monitor" becomes "measure", for example), it is always safer to use quotes. The ID can contain a wildcard, such as measure ID "m*" off; measure ID "*3" on;. text, font, alignment, radius, and color can be changed independently once a measurement with ID has been defined. Options FONT, ALIGN, RADIUS, and COLOR are shown here separately, but they could be given in a single command, in any order. Thus: MEASURE ID "m1" "test" @3 @7 font 15 sansserif bold align left radius 0.1 and, later, MEASURE ID "m1" color red radius 0.1.','0|1|2|3|4','','') newCmd('','','','*v+13.0;','Specify the font size (10,15,20), name (SERIF, SANSSERIF), and weight (BOLD,PLAIN,ITALIC).','*2','"id" "label"','FONT','size name face','') newCmd('','','','*v+13.0;','Specify the radius of the dots used to show this measurement. RADIUS 0.0 results in no dots. ','*3','"id" "label"','RADIUS','(decimal)','') newCmd('','','','*v+13.0;','Specify the text alignment of the label.','*1','"id" "label"','ALIGN','LEFT/RIGHT/CENTER','') newCmd('','','','*v+13.0;','Turn on or off a measurement or measurement group, or delete it. ','*5','"id" "label"','ON/OFF/DELETE') newCmd('','','','*v+13.0;','Specify the color for this measurement','*4','"id" "label"','COLOR','._colorRGB','') newCmd('.menu','13.0.RC4','','*v+13.0 -- new;','Accepted only as the sole contents of a script, the menu command pops up the context menu for the user as though they had right-clicked the applet or clicked on the Jmol logo. JSmol note : The JavaScript setting Jmol._persistentMenu = true allows menu to persist and not be removed when the mouse is moved off of it.','1','','') newCmd('.meshribbon','','structure','disp','A mesh ribbon is similar to a strand, but is more the quality of a loosely woven fabric. ','0|1','','') newCmd('','','','','','*11','._on_off{"ON"}','') newCmd('','','','*v+11.6;','Turns meshribbon rendering on and all other rendering (including labels) off.','*12','ONLY','') newCmd('','','','','A negative number also implies ONLY.','*2','._mesh_ribbon_radius','') newCmd('.message','','','*v+11.0 -- NEW;math','Sends a string of text to the messageCallback function (for the applet). The {#.set_MessageCallback~} command can be used to set this JavaScript function. The message is also entered into the scriptStatus queue. Variable values and math expressions can be included in messages and echos. These use the following syntax: message my variable = @{{variablename}}, provided a variable with that name exits. Note that the braces are required -- message my variable = @x will simply deliver "my variable = @x". The @{{...}} syntax is also supported in this context: message the fraction 3 / 2 is @{{ 3.0 / 2 }}. Basically, any expression that can appear in a {#.print~} command can appear in @{{...}} in a message or echo. For example: message @{{ {{_N and connected(2,_H)}}.size }} NH2 groups are present. Since the Jmol application can be run "headless" -- with no display -- using the -ions set of flags, you can use a designed message command to deliver model information to other programs, however the {#.print~} command has a simpler syntax and is more flexible.','1','(string)','') newCmd('.minimize','','','*v+13.0 -- replaces the default UFF calculation with MMFF94, where applicable.;','Minimizes the structure using the MMFF94 force field (T. A. Halgren, J. Comp. Chem. 5 & 6, 616-641 (1996) or an adapted UFF force field (Rappe, A. K., et. al.; J. Am. Chem. Soc. (1992) 114(25) p. 10024-10035). The default force field is MMFF94, but the UFF force field will automatically be used if atoms are not fo types recognized by MMFF94 or if set forceField "UFF" is issued. enerally the minimization runs in its own thread. If it is desired to wait until the process has completed prior to continuing, use set useMinimizationThread false. You can retrieve a detailed report of the minimization energy using the "high debug" setting and then either show minimization or x = script("show minimization");print x:
set loglevel 6
minimize steps 0
x = script("show minimization")
set debug off
print x
Minimization always runs on the currently displayed frame or set of frames. Generally, this allows selective minimization of the currently displayed model. If sequential minimization of multiple models independently is desired, one can do this two ways:
(1) Issue a {#.frame~} command to call up the desired model, then issue MINIMIZE;
(2) Make visible all models using FRAME *, and then selectively minimize just the models you want to minimize.
The following two scripts minimize all models in a collection sequentially and independently:
set useMinimizationThread false
var n = {{*}}.model.max
for (var i = 1; i <= n; i++) {{:> frame @i:> minimize
}}
set useMinimizationThread false
var n = {{*}}.model.max
frame *
for (var i = 1; i <= n; i++) {{:> minimize {{model=i}}
}}
Note that in either case we must use {#.set_useminimizationThread~set useMinimizationThread FALSE} because we are doing sequential minimizations within a script.','0','','') newCmd('','','','*v+11.6;','Carries out a minimization using the default convergence criterion specified by {#.set(misc)~set minimizationCriterion} or until the maximum number of steps specified by {#.set(misc)~set minimizationSteps} is reached. If {#.set(misc)~set minimizationRefresh} is TRUE, then the model refreshes after each minimization step, producing a sort of animated minimization.','*10','','') newCmd('','13.3.9','','*v+14.0;','Carries out a minimization using the specified atom set and assumes the SELECT and ONLY options, thus ignoring all other atoms. Bonds to atoms connected to this set are ignored and may lengthen or shorten during the minimization.','*11','._atom_expression','') newCmd('','11.9.19','','*v+12.0;','First adds hydrogen atoms, then minimizes the structure.','*2','ADDHYDROGENS','') newCmd('','','','','Stops a running minimization.','*3','CANCEL','') newCmd('','11.5.24','','','Stops a running minimization and clears all constraints ','*4','CLEAR','') newCmd('','11.5.24','','*v+11.6;','Clears any current constraints.','*5','CONSTRAINT','CLEAR') newCmd('','11.5.24','','*v+11.6;','Constrains the distance, angle, or dihedral angle invoving two to four atoms. If a given atom expression involves more than one atom, only the first atom in the set is made part of the constraint.','*6','CONSTRAINT','(two to four atom expressions)','(decimal)','') newCmd('','11.5.23','','*v+11.6;','Overrides the setting of {#.set_minimizationCriterion~minimizationCriterion} for this minimization.','*7','CRITERION','') newCmd('','11.5.24','','*v+11.6;','Do no minimization -- just calculate the energy','*8','ENERGY','') newCmd('','11.5.24','','*v+11.6;','Specifies a set of atoms to remain in fixed positions during this minimization and future minimizations (if additional parameters are present) or only in future minimizations (if not). Use minimize fix NONE to clear this setting.','*9','FIX','._atom_expression') newCmd('','13.3.9','*v+14.0','*v+14.0;','Minimizes only the specified atom set. Any previous or additional setting of FIX is respected. If FIX is not explicitly given, the specified atoms are minimized by fixing all other atoms in the molecule and then internally setting the set of atoms to minimize to be ALL. ','*911','SELECT','._atom_expression') newCmd('','16.1.13','','*v+16.1;','Carries out a minimization using the specified group of atoms, fixing all other atoms. The positions of any atoms in the group that are bonded to atoms outside this set are also fixed, such that bonds only within the group are changed. Note that the UFF forcefield will likely be used, because the resulting subgroup of atoms may be only a fragment of a molecule.','*910','GROUP','._atom_expression') newCmd('','13.3.9','','*v+14.0;','Minimizes only the specified atom set, specifically ignoring all other atoms, even if they are connected to the selected atoms. Note that the UFF forcefield will likely be used, because the atom set may be only a fragment of a molecule.','*915','SELECT','._atom_expression','ONLY','') newCmd('','','','*v+11.6;','Overrides the setting of {#.set(misc)~minimizationSteps} for this calculation. ','*92','STEPS','(integer)') newCmd('','','','','Same as CANCEL.','*93','STOP','') newCmd('.mo','','','*v+11.2 -- MO SLAB, MO POINTS;iso','
 The MO (molecular orbital) command displays molecular orbitals contained in a variety of file formats. One simply loads the file, then selects which orbital to display with MO n where "n" is the number of the molecular orbital to display. (With some file formats, you may need to use the {#.frame~frame or model} command to call up the specific model having the MOs.) Option changes take effect immediately with the currently displayed orbital and stay in effect for later MO commands until a new file is loaded.
The MO (molecular orbital) command displays molecular orbitals contained in a variety of file formats. One simply loads the file, then selects which orbital to display with MO n where "n" is the number of the molecular orbital to display. (With some file formats, you may need to use the {#.frame~frame or model} command to call up the specific model having the MOs.) Option changes take effect immediately with the currently displayed orbital and stay in effect for later MO commands until a new file is loaded. Note that ONE molecular orbital is allowed per model (that is, per frame). The {#.isosurface~} command can be used for more advanced molecular orbital display options or for displaying several planes or orbitals simultaneously. The default rendering is MESH NOFILL FRONTONLY. Where indicated below, adding the SQUARED keyword generates the linear combination of all degenerate orbitals if one is indicated (by a number, or HOMO, LUMO, NEXT, or PREV) or the specified set of orbitals in the case of a linear combination, squaring the wave functions prior to adding. In general, this results in showing the contribution of this set of orbitals to the overall total electron density and the symmetry of that density.','1|2','','') newCmd('','','','*v+11.0;','Turn on/off the molecular orbital.','*10','._on_off{"ON"}','') newCmd('','','','*v+11.0;','Selects the specific molecular orbital to dispay, starting with the number 1. If this number is negative, the colors of the phases are reversed.','*310','(integer)','[SQUARED]') newCmd('','13.1.9','','*v+11.0;','Calculates the sum of the squares of all occupied wave functions, effectively showing overall electron density.','*311','DENSITY','') newCmd('','','','*v+12.2;','Displays a linear combination of molecular orbitals based on a set of coefficients and orbital numbers, for example: mo [0.5, 20, 0.5, 21]. Coefficients will automatically be normalized such that SUMi(ci*ci) = 1.00. If variables are involved, one needs @{{...}}. Thus, load c6h6.smol; for (f = 0; f <= 10; f++) {{ mo @{{[f/10.0, 20, (10-f)/10.0, 21]}}; refresh }}. Note that this allows creation of a reversed-phase orbital using -1.0 for the coefficient of a single orbital: mo [-1.0 28].','*32','[c1 n1,c2 n2, c3 n3,...]','[SQUARED]') newCmd('','','','*v+11.2;','Selects the highest doubly- or singly-occupied molecular orbital, optionally an orbital +/-n from this orbital. (Only applicable to data sets that have orbital occupancies indicated in the file.) ','*61','HOMO','[+/-n]','[SQUARED]','') newCmd('','','','*v+11.2;','Selects the lowest unoccupied molecular orbital, optionally an orbital +/-n from this orbital. (Only applicable to data sets that have orbital occupancies indicated in the file.)','*62','LUMO','[+/-n]','[SQUARED]','') newCmd('','12.0.RC6','','*v+12.0;','Specifies the model for this command; must be followed by an orbital specification such as HOMO or 12.','*64','MODEL','n or x.y') newCmd('','','','*v+11.0;','Displays the next MO in the file using the same characteristics as the currently displayed one.','*65','NEXT','[SQUARED]') newCmd('','','','*v+11.0;','Displays the previous MO in the file using the same characteristics as the currently displayed one.','*85','PREVIOUS','[SQUARED]') newCmd('','','','*v+11.0;','Goes back to full orbital display rather than a planar slice.','*68','NOPLANE','') newCmd('','','','*v+11.0;','Indicates that what is desired is a color-mapped planar slice through the orbital using one of the ways of expressing a {#.plane expressions~plane}. ','*80','PLANE','plane_expression') newCmd('','12.1.48','','*v+12.2;','Produces a "pointilist" probability-based visualization of a molecular orbital using the specified number of points and an optional random seed. ','*81','POINTS','numberOfPoints','randomSeed','') newCmd('','','','*v+12.2;','Modifies the scale of the box used to sample the molecular orbital. Note that the SCALE option does not change the size of the orbital. The default scale of 1.0 uses an algorithm that generally produces integrations of 1.000 +/- 0.001, indicating sufficient sampling size and grid resolution. But some cases involving very diffuse orbitals may require larger scales. Adjustment of this parameter may also require changes to the MO RESOLUTION parameter. (An increase in scale may also require an increase in resolution to achieve acceptable integration.)','*82','SCALE','(decimal){1.0}') newCmd('','','','*v+11.0;','Colors the orbital one specific color','*40','COLOR','._colorRGB') newCmd('','','','*v+11.0;','Colors regions where the wave function is less than zero the first color; regions where the wavefunction is greater than zero the second color.','*51','COLOR','._colorRGB','._colorRGB','') newCmd('','','','*v+11.0;','Delete the molecular orbital.','*60','DELETE','') newCmd('','','','*v+11.0;','Sets the cutoff value for the isosurface that defines the orbital. This number may be dependent upon the computational package used to generate the orbitals. Values in the range 0.005 - 0.05 may need to be experimented with in order to get the best display. Values closer to zero lead to surfaces further from the atoms (larger orbitals). Both positive and negative cutoffs are allowed. A positive number indicates to use both positive and negative cutoffs. Adding an explicit "+" sign before the number indicates that only the positive part of the surface is desired.','*55','CUTOFF','(decimal)') newCmd('','','','*v+11.0;','Sets the resolution of the isosurface rendering in "points per Angstrom". Higher resolution leads to smoother surfaces and more detail but carries the penalty of slower surface generation. Typical values for molecular orbitals are 4-10 points per Angstrom.','*92','RESOLUTION','(decimal)') newCmd('','12.1.50','','*v+12.2;','MO SLAB provides an extensive mechanism allowing for the trimming of a molecular orbital using a wide variety of methods. MOs can be "slabbed" based on planes, spheres, bounding boxes, unit cells, proximity to a set of atoms, within a range of values, or a dynamic z-plane relative to the user. The section removed can be removed completely or, using MO SLAB TRANSLUCENT, left as a translucent "ghost" surface. An MO may be slabbed any number of times in any number of ways. The slabbing is reversible using MO SLAB NONE for general slabbing, and using MO SLAB OFF for dynamic z-plane slabbing. Options include: [|| {#.[plane expression]~(plane expression)} | a plane. Note that plane definitions starting with "-" switch sides of the plane ||BOUNDBOX | within the currently defined {#.boundbox~bounding box} ||UNITCELL | within the currently defined {#.unitcell~unit cell} ||WITHIN x.y {{atom expression}} | within the given distance of the specified atom set ||WITHIN x.y (point) | within the given distance of the specified point (a spherical slab) ||WITHIN RANGE x.x y.y | within the value range x.x and y.y. ||]','*93','SLAB','(parameters)') newCmd('','','','*v+11.0;','Sets the format of the orbital title appearing in the upper left corner of the applet. Special format characters include:
[|| %E or %0.dE|energy (possibly rounded to d decimal digits||%F|filename || %I | molecular orbital number || %M | model number || %N | total number of molecular orbitals || %O|occupancy ||%S|symmetry||%U|energy units|| | | (vertical bar) new line|| ? |(at the beginning of a line) indicates to disregard line if no data for that line are present in the file||]
If a formatted item is not indicated in the file, then it is left blank. The default title is "%F | Model %M MO %I/%N | Energy = %E %U | ?Symmetry = %S | ?Occupancy = %O". The command MO titleFormat "" may be used to show no title. ','*95','TITLEFORMAT','"format"') newCmd('.model','14.1.1','','*v+14.2;model','The MODEL command functions the same as the {#.frame~} command with one exception: In the case of model n where n is an integer, and a single PDB file containing MODEL records is loaded, the integer used here refers to the number specified in a MODEL record, not the model\'s sequential position in the file. ','1|1','','') newCmd('','14.1.1','','*v+14.2;','In the case of the loading of a single PDB file containing MODEL records, the integer used here corresponds to the number in that record, so model 1 does not necessarily load the first model in the file, the way frame 1 would. ','*1','(integer)','') newCmd('','14.3.15','','*v+14.4;','This command adds property data to model\'s auxilliaryInfo. x may be any valid math expression. For example, model 1 property "test" {{1.1}}.temperature.mul(0.1) creates _M.test, which is an array. If the source variable is an array, then the data can also be used to create atom data: {{1.1}}.property_t = {{1.1}}.label("%{{test}}"). ','*2','(integer)','PROPERTY','name','(list)') newCmd('','','','*v+14.4;','Loads DSSR data from a file.','*3','(integer)','PROPERTY','DSSR','"filename"') newCmd('','','','*v+14.4;','Loads DSSR data from a variable.','*4','(integer)','PROPERTY','DSSR','@v') newCmd('.modelkit','14.32.2','','*v+14.32;','The MODELKIT command (introduced in Jmol 14.32.2) allows scripted access to the Jmol ModelKit without using {#.set_modelkitmode~set modelkitMode}. modelkit ASSIGN and modelkit CONNECT can be used to build or change a any model. modelkit MUTATE modifies the residue structure of proteins and alllows the construction of simple peptides with defined secondary structure. Modelkit options can be set using modelkit SET followed by a modelkit configuration option. This command is in development and is not fully documented.','1|1','','') newCmd('','','','*v+14.32;','Assign an atom a new element.','*11','ASSIGN ATOM','{{atom}}','"element"','') newCmd('','','','','Connect two atoms.','*13','CONNECT','{{atom}}','{{atom}}','') newCmd('','14.32.7','','*v+14.32;','Create a peptide with a specified sequence and secondary structure motif (various alpha-, beta-, and turn-forms) based on phi/psi angles. See the {#.mutate~} command for details.','*71','MUTATE','CREATE','options','') newCmd('','14.32.7','','*v+14.32;','Mutate a specific protein residues. See the {#.mutate~} command for details.','*72','MUTATE','(integer)','options','') newCmd('','14.32.7','','*v+14.32;','Mutate one or more protein residues. See the {#.mutate~} command for details.','*73','MUTATE','{{residues}}','options','') newCmd('.modulation','13.3.4','','*v+14.0;','The MODULATION command, introduced in Jmol 14.0, allows visualization of {http://www.ccp14.ac.uk/ccp/web-mirrors/crys-methods/Project/modulate/modulate.html~incommensurately modulated strutures} that have been read from msCIF or JANA2006 files. An (nonrealistic) dynamic phased animation of the structure can be activated using {#.vibration~vibration ON}, with the magnitude of the oscillations governed by {#.set_modulationScale~set modulationScale}. Note that incommensurately modulated structures are never actually oscillating in such a way; different t values actually correspond to different physical unit cell positions in the crystal. This animation only serves as a visualization of the phasing correlations in a given unit cell.','1|1','','') newCmd('','13.3.4','','*v+14.0;','Turns modulation on, moving atoms to their modulated positions and displaying the aperiodic structure.','*10','ON','') newCmd('','13.3.4','','*v+14.0;','Turns modulation off, displaying the unmodulated, periodic structure.','*20','OFF','') newCmd('','14.0.1','','*v+14.0;','Turns modulation on or off for a specific set of atoms only, adding or removing those atoms to/from the set of atoms already modulated.','*21','._atom_expression','ON/OFF') newCmd('','14.0.1','','*v+14.0;','Specifies the parameter t, the coordinate along the hyperspace axis, setting the phase of the modulations and creating a projection of the (3+n)D structure in 3D. If n > 1, sets all hyperspace coordinates to t. Values range from 0.0 to 1.0. Note that using the integer 1 is NOT the same as using the number 1.0, as integers are used to give multiples of q (see below).','*3','T','(decimal)') newCmd('','14.0.1','','*v+14.0;','Specifies distinct phase settings for up to three wave vectors. Values range from 0 to 1.','*4','T','{{t1 t2 t3}}') newCmd('','13.3.4','','*v+14.0;','Specifies the parameter t as multiples of the wave vector q, the coordinate along the hyperspace axis, setting the phase of the modulations and creating a projection of the (3+n)D structure in 3D. If the modulation dimension n > 1, sets all hyperspace coordinates to m times the length of the wave vector . Values must be integers. The effect of this setting is to shift the displayed unit cell by m unit cells along the modulation vector.','*5','Q','(integer)') newCmd('','14.0.1','','*v+14.0;','Specifies distinct phase settings for up to three wave vectors. The second parameter TRUE distinguishes this from setting t values directly.','*6','Q','{{m1 m2 m3}}') newCmd('','14.0.1','','*v+14.0;','Sets the scale of all modulations.','*7','scale','x.x') newCmd('.move','','','anim;camera','The move command provides powerful animation capabilities. It effectively changes the camera position, allowing you to specify rotations, zooming, and translations to be performed in a specified period of time. xRot, yRot, and zRot are rotations about the cartesian axes in degrees. Zoom specifies a zoom factor, xTrans, yTrans, and zTrans are translations in the range -100 to 100. If you do not know what slab is, just put in a zero. see the slab command for more information. This command has been superceded by the moveTo command.','11','._drotx','._droty','._drotz','._dzoom;+._dtransx;+._dtransy;+._dtransz;+._dSlab;+._floatSecondsTotal;+._move_fps{30};+._move_maxAccel{5}') newCmd('.moveto','','','*v+11.2 -- adds navigation capability;anim;nav;camera','
 The moveto command rotates the molecule to a predefined orientation, as though the camera were moving smoothly to a new position. Two formats can be used. In each, the first (optional) parameter specifies the number of seconds during which the molecule should rotate smoothly from the current orientation to the new orientation. A 0 for this first parameter specifies an instantaneous reorientation. If the axis is {{0 0 0}} and the degrees are 0, then the molecule is not reoriented during the operation. In conjunction with "show orientation" this command allows reading and restoring specific user-specified orientations.','1|2','','')
newCmd('','','','*v+11.0;','A simple use of moveTo just has six optional directions. ','*3','timeSeconds','FRONT|BACK|LEFT|RIGHT|TOP|BOTTOM')
newCmd('','','','*v+11.2;','In the second option, the second parameter is a coordinate {{x, y, z}} defining the axis relative to the default orientation about which the molecule should be rotated. The third parameter is the counterclockwise (right-hand) rotation in degrees about this axis. "moveto 0 {{0 0 1}} 0" rotates the model to the default orientation (equivalent to "reset"). If the angle parameter is 0 but any one of x, y, or z is nonzero, then no reorientation occurs (because the axis has been specified, but the rotation is 0 degrees). Following these parameters is the zoom setting in percent, the X- and Y-positions of the rotation center on the screen, as percent of width and height, respectively. The actual molecular coordinate of the rotation center along with the rotation radius (which determines the magnification associated with ZOOM 100) are next. The final parameters define the navigation center molecular coordinate, its X- and Y- position on the screen in percent, the depth of the navigation point in percent of model depth (100 = front, 0 = rear), camera depth, and camera x and y position. In conjunction with "show/save/restore orientation" this command allows reading and restoring specific user-specified orientations.','*4','timeSeconds','{{x y z}}','degrees','zoomPercent;+transX;+transY;+{{x y z}};+rotationRadius;+navigationCenter;+navTransX;+navTransY;+navDepth;+cameraDepth;+cameraX;+cameraY')
newCmd('','11.1.36','','*v+11.2;','If the zoom setting prior to translation positions is 0, and an atom expression is used for the point, then the moveTo can be designed to automatically zoom to the scale that would fill the screen with that set of atoms. The optional zoom adjustment is in the form +n, -n, *n, or /n, as for {#.zoomTo~}.','*6','timeSeconds','{{x y z}}','degrees','0;+transX;+transY;+(atom expression);+0;+zoomAdjustment;+navigationCenter;+navTransX;+navTransY;+navDepth;+cameraDepth;+cameraX;+cameraY')
newCmd('','11.1.36','','*v+11.2;','If no translation is involved, then there is also no need for the zoom setting of 0 prior to the atom expression.','*7','timeSeconds','{{x y z}}','degrees','(atom expression);+0;+zoomAdjustment;+navigationCenter;+navTransX;+navTransY;+navDepth;+cameraDepth;+cameraX;+cameraY')
newCmd('','11.9.8','','*v+11.2;','Reorients the model to the orientation specified by the quaternion given in {{x y z w}} format. ','*80','timeSeconds','QUATERNION','{{ quaternion }}','')
newCmd('','11.9.8','','*v+11.2;','Reorients the model to the orientation specified by the quaternion specified by the atom expression and the current setting of quaternionFrame.','*81','timeSeconds','QUATERNION','._atom_expression','')
newCmd('','11.9.8','','*v+11.2;','Reorients the model to the orientation specified by a molecular frame quaternion, as, for example, retrieved by q = quaternion({{resno=30}}) and then used in moveto quaternion MOLECULAR @q. Note that because this quaternion refers to the rotation required to transform the reference frame to the specified molecular frame, in order to "move to" this orientation, one must use its inverse (bring the molecular frame to the reference frame, q_orientation = !q_molecular). The keyword MOLECULAR simply applies this inversion. ','*82','timeSeconds','QUATERNION','MOLECULAR','{{ quaternion }}')
newCmd('','14.1.16','','*v+14.6;','Reorients the viewpoint to de down one of the crystallographic axes (a, b, or c), positioning that axis one of the following positions: (1) upper left, (2) upper right, (3) lower right, (4) lower left. Adding a minus sign points the axis away from the viewer rather than toward the viewer.','*21','timeSeconds','AXIS','[a1,b1,c1,a2,b2,c2,a3,b3,c3,a4,b4,c4,-a1,-b1,-c1,-a2,-b2,-c2,-a3,-b3,-c3,-a4,-b4,-c4]','')
newCmd('','14.1.16','','*v+14.2;','Reorients the viewpoint to de down a specific axis, either one of the crystallographic axes (a, b, or c), or one of the Cartesian axes (x, y, z). "a" is shorthand for "a1", "b" for "b1", and "c" for "c4".','*22','timeSeconds','AXIS','[a,b,c,x,y,z]','')
newCmd('','11.9.24','','*v+11.2;','Stops an asychronous moveto operation that has been started after setting set waitForMoveTo FALSE. ','*8','STOP','')
newCmd('.mutate','14.3.13','','*v+14.4;','The MUTATE command is used to replace one or more amino acids in the current model frame by another or to create polypeptides in a new frame with defined phi and psi angles. (See {#.mutate (CREATE)~mutate_CREATE}.) Jmol flexible fit using Jmol bioSMARTS (x = compare(res1,res0, "[*.N][*.CA][*.C][*.O]", "BONDS"); rotate branch @x 1) is used to set the backbone dihedral angle. Similarly, Jmol bioSMARTS is used to move atoms to their proper location (compare res1 res0 SMARTS "[*.N][*.CA][*.C]" rotate translate 0). Then, necessary atoms are removed and bonds are made. No attempt is made to optimize the side-chain conformation. However, using set picking dragMinimize, one can quickly reorient the side-chain to a more appropriate conformation if desired. The mutated structure can be saved using the {#.write~write PDB} option. The general format for the MUTATE command is:
The moveto command rotates the molecule to a predefined orientation, as though the camera were moving smoothly to a new position. Two formats can be used. In each, the first (optional) parameter specifies the number of seconds during which the molecule should rotate smoothly from the current orientation to the new orientation. A 0 for this first parameter specifies an instantaneous reorientation. If the axis is {{0 0 0}} and the degrees are 0, then the molecule is not reoriented during the operation. In conjunction with "show orientation" this command allows reading and restoring specific user-specified orientations.','1|2','','')
newCmd('','','','*v+11.0;','A simple use of moveTo just has six optional directions. ','*3','timeSeconds','FRONT|BACK|LEFT|RIGHT|TOP|BOTTOM')
newCmd('','','','*v+11.2;','In the second option, the second parameter is a coordinate {{x, y, z}} defining the axis relative to the default orientation about which the molecule should be rotated. The third parameter is the counterclockwise (right-hand) rotation in degrees about this axis. "moveto 0 {{0 0 1}} 0" rotates the model to the default orientation (equivalent to "reset"). If the angle parameter is 0 but any one of x, y, or z is nonzero, then no reorientation occurs (because the axis has been specified, but the rotation is 0 degrees). Following these parameters is the zoom setting in percent, the X- and Y-positions of the rotation center on the screen, as percent of width and height, respectively. The actual molecular coordinate of the rotation center along with the rotation radius (which determines the magnification associated with ZOOM 100) are next. The final parameters define the navigation center molecular coordinate, its X- and Y- position on the screen in percent, the depth of the navigation point in percent of model depth (100 = front, 0 = rear), camera depth, and camera x and y position. In conjunction with "show/save/restore orientation" this command allows reading and restoring specific user-specified orientations.','*4','timeSeconds','{{x y z}}','degrees','zoomPercent;+transX;+transY;+{{x y z}};+rotationRadius;+navigationCenter;+navTransX;+navTransY;+navDepth;+cameraDepth;+cameraX;+cameraY')
newCmd('','11.1.36','','*v+11.2;','If the zoom setting prior to translation positions is 0, and an atom expression is used for the point, then the moveTo can be designed to automatically zoom to the scale that would fill the screen with that set of atoms. The optional zoom adjustment is in the form +n, -n, *n, or /n, as for {#.zoomTo~}.','*6','timeSeconds','{{x y z}}','degrees','0;+transX;+transY;+(atom expression);+0;+zoomAdjustment;+navigationCenter;+navTransX;+navTransY;+navDepth;+cameraDepth;+cameraX;+cameraY')
newCmd('','11.1.36','','*v+11.2;','If no translation is involved, then there is also no need for the zoom setting of 0 prior to the atom expression.','*7','timeSeconds','{{x y z}}','degrees','(atom expression);+0;+zoomAdjustment;+navigationCenter;+navTransX;+navTransY;+navDepth;+cameraDepth;+cameraX;+cameraY')
newCmd('','11.9.8','','*v+11.2;','Reorients the model to the orientation specified by the quaternion given in {{x y z w}} format. ','*80','timeSeconds','QUATERNION','{{ quaternion }}','')
newCmd('','11.9.8','','*v+11.2;','Reorients the model to the orientation specified by the quaternion specified by the atom expression and the current setting of quaternionFrame.','*81','timeSeconds','QUATERNION','._atom_expression','')
newCmd('','11.9.8','','*v+11.2;','Reorients the model to the orientation specified by a molecular frame quaternion, as, for example, retrieved by q = quaternion({{resno=30}}) and then used in moveto quaternion MOLECULAR @q. Note that because this quaternion refers to the rotation required to transform the reference frame to the specified molecular frame, in order to "move to" this orientation, one must use its inverse (bring the molecular frame to the reference frame, q_orientation = !q_molecular). The keyword MOLECULAR simply applies this inversion. ','*82','timeSeconds','QUATERNION','MOLECULAR','{{ quaternion }}')
newCmd('','14.1.16','','*v+14.6;','Reorients the viewpoint to de down one of the crystallographic axes (a, b, or c), positioning that axis one of the following positions: (1) upper left, (2) upper right, (3) lower right, (4) lower left. Adding a minus sign points the axis away from the viewer rather than toward the viewer.','*21','timeSeconds','AXIS','[a1,b1,c1,a2,b2,c2,a3,b3,c3,a4,b4,c4,-a1,-b1,-c1,-a2,-b2,-c2,-a3,-b3,-c3,-a4,-b4,-c4]','')
newCmd('','14.1.16','','*v+14.2;','Reorients the viewpoint to de down a specific axis, either one of the crystallographic axes (a, b, or c), or one of the Cartesian axes (x, y, z). "a" is shorthand for "a1", "b" for "b1", and "c" for "c4".','*22','timeSeconds','AXIS','[a,b,c,x,y,z]','')
newCmd('','11.9.24','','*v+11.2;','Stops an asychronous moveto operation that has been started after setting set waitForMoveTo FALSE. ','*8','STOP','')
newCmd('.mutate','14.3.13','','*v+14.4;','The MUTATE command is used to replace one or more amino acids in the current model frame by another or to create polypeptides in a new frame with defined phi and psi angles. (See {#.mutate (CREATE)~mutate_CREATE}.) Jmol flexible fit using Jmol bioSMARTS (x = compare(res1,res0, "[*.N][*.CA][*.C][*.O]", "BONDS"); rotate branch @x 1) is used to set the backbone dihedral angle. Similarly, Jmol bioSMARTS is used to move atoms to their proper location (compare res1 res0 SMARTS "[*.N][*.CA][*.C]" rotate translate 0). Then, necessary atoms are removed and bonds are made. No attempt is made to optimize the side-chain conformation. However, using set picking dragMinimize, one can quickly reorient the side-chain to a more appropriate conformation if desired. The mutated structure can be saved using the {#.write~write PDB} option. The general format for the MUTATE command is:mutate [target residue(s)] [amino acid or sequence]
Target residues may be indicated as an integer residue number, mutate 33..., a standard XXXnn mutation code, mutate ALA34..., or any atom expression that specifies one or more residues, mutate {{1-3}} ....
Amino acids may be indicated with their three-letter code (for example, mutate 33 LYS, loaded as "==LYS", from RCSB), a standard one-letter standard sequence code (for example, mutate ASP34 K), or as a filename in quotes: mutate 33 "myLYS.pdb".
Sequences can be given using single-letter or three-letter codes. In the unusual case where the three single letters spell a three-letter code, such as LYS or ALA, they must be prefixed with a tilde: ˜LYS, ˜ALA. When more than one target residue is indicated and a shorter sequence is given, the sequence will repeated as necessary to change all the residues. For example: mutate {{1-4}} "ALA-VAL" will suffice for "ALA-VAL-ALA-VAL", and mutate {{1-3}} V is equivalent to mutate {{1-3}} "VAL-VAL-VAL". The underscore character in a sequence means "no change". Similarly, for three-letter codes, use "UNK" to skip a residue.
Examples are given below:
[||mutate 33 LYS| Change residue 33 to lysine.||mutate ALA34 G| Change residue 34 to glycine.||mutate {{ALA.CA}}[2] G|Change only the second ALA residue to glycine. Note the use of ".CA" to create a list of residues, then [2] to select the second of those.||mutate {{1-3}} GAL|Change the first three residues to GLY-ALA-LEU. ||mutate {{1-3}} A| Change the first three residues to alanine. The specified sequence will be repreated as necessary to cover the indicated residues.||mutate {{1-3}} ˜LYS| Change the first three residues to LEU-TYR-SER. Tilde is required in this case ony because the sequence spells the three-letter code for the amino acid lysine. ||mutate {{1-3}} G_L|Change the first residue to glycine and the third residue to leucine, making no change at the second.||mutate {{1-4}} "GLY-AlA"|Modify the first four residues using 3-letter amino acid codes. The sequence in this case will be GLY-ALA-GLY-ALA. (Note that quotations are required for three-letter sequences because of the use of the hyphen.) ||mutate ALA34 "myAla.pdb"|Change ALA34 to the amino acid to be found in the single-residue PDB format file "myAla.pdb". ||mutate ALA34 "==SEP"|Specify a non-standard PDB component using "==" and the three-letter PDB code for the component, as for the {#.load~} command. In this case, we are specifying phopshoserine. ||mutate @3 @res|Variables are allowed for target or sequence.||]
','1|2','','') newCmd('.mutate (CREATE)','14.32.7','','*v+14.32;','MUTATE CREATE creates a polypeptide in a new frame with a specified secondary structure using the same syntax as for {#.mutate~direct mutation}. The general format for the MUTATE CREATE option is:
mutate CREATE [sequence] [secondary structure specification]
Sequences can be given as described for {#.mutate~} using single-letter or three-letter codes.
Secondary structure can be given using an array of phi/psi pairs, which will be replicated as necessary to complete the sequence. For example: mutate CREATE aaaaaaaa [-65, -40] creates a standard alpha helix of eight alanine residues, and mutate CREATE gggggggg "beta" generates an all-glyciine beta strand. Predefined motifs are given below and could easily be expanded upon request. They are not case-sensitive: [|| "alpha"| [ -65, -40 ]|idealized alpha helix || "3-10"| [ -74, -4 ] | 310 helix || "pi"| [ -57.1, -69.7 ] | idealized pi helix || "alpha-L"| [ 57.1, 4 ] | left-handed alpha helix || "helix-II"|[ -79, 150 ] | proline Type-II helix|| "collagen"| [ -51, 153 ] |collagen wide-coil helix allowing for triple helix || "beta"| [ -140, 130 ] | slightly twisted beta strand || "beta-120"| [ -120, 120 ] | 120-degree beta strand|| "beta-135"| [ -135, 135 ] |perpendicular side-chain beta strand || "extended"| [ 180, 180 ] | fully extended chain || "turn-I"| [ -60, -30, -90, 0 ] | turn type I || "turn-II"| [ -60, 120, 80, 0 ] | turn type II || "turn-III"| [ -60, -30, -60, -30 ] | turn type III || "turn-I\'"| [ 60, 30, 90, 0 ] | turn type I\' || "turn-II\'"| [ 60, -120, -80, 0 ] | turn type II\' || "turn-III\'"| [ 60, 30, 60, 30 ] | turn type III\' ||] ','1|2','','') newCmd('.navigate','11.1.1','','*v+11.2 -- new;nav','The navigate command allows exploring of the model from the perspective of an airplane capable of flying through the model. In each case, an optional time in seconds determines the time to reach the objective. If no time is given, the change occurs over two seconds. (In the case of PATH, this is the time to each point along the path, not the total path.) Subcommands can be grouped in the same command by separating them with "/", for example: navigate 2 DEPTH 30 / 5 ROTATE 180 / DEPTH 20 / TRANSLATE X 10.','0|1','','') newCmd('','11.1.1','','*v+11.2;','Bring the observer to the specifed molecular coordinate, which may be fractional in the case of crystal structures.','*11','timeSeconds','CENTER','{{x y z}}','') newCmd('','11.1.1','','*v+11.2;','Bring the observer to the geometric center of the set of atoms specified in parentheses.','*12','timeSeconds','CENTER','._atom_expression','') newCmd('','11.1.1','','*v+11.2;','Bring the observer to the geometric center of the points corresponding to the indicated {#.draw} object.','*13','timeSeconds','CENTER','$object','') newCmd('','11.1.1','','*v+11.2;','Bring the observer forward or backward to the depth indicated as a percent from back (0) to front (100) of the model. Values can be negative, in which case until a rotate command is given, only screen background will be seen. Values greater than 100 put the observer outside the model. ','*20','timeSeconds','DEPTH','percent','') newCmd('','11.1.1','','*v+11.2;','Follows the Hermite path given by the specified {#.draw~} object (such as a line, curve, or arrow). This allows easy dynamic development of paths using a predefined draw object followed by set picking draw and show draw. ','*32','timeSeconds','PATH','$object','') newCmd('','11.1.1','','*v+11.2;','Follows a Hermite path defined by a set of nodes expressed in terms of any combination of atom sets, draw objects, or coordinates.','*33','timeSeconds','PATH','(any combination of coordinates, atom expressions, and objects)','') newCmd('','11.1.1','','*v+11.2;','Rotates around the X axis at the navigation center.','*42','timeSeconds','ROTATE','X','degrees') newCmd('','11.1.1','','*v+11.2;','Rotates around the Y axis at the navigation center.','*44','timeSeconds','ROTATE','Y','degrees') newCmd('','11.1.1','','*v+11.2;','Rotates around the Z axis at the navigation center.','*46','timeSeconds','ROTATE','Z','degrees') newCmd('','11.1.1','','*v+11.2;','Navigates along the trace of a protein or nucleic acid. The "ride" can be changed depending upon the settings of {#.set(structure)~set sheetsmoothing} and {#.set(structure)~set tracealpha}.','*61','timeSeconds','TRACE','._atom_expression','') newCmd('','11.1.1','','*v+11.2;','Translates the navigation screen offset to the specified positions expressed as percent of width (X) and height (Y) of the applet/application window.','*62','timeSeconds','TRANSLATE','x.xx','y.yy') newCmd('','11.1.1','','*v+11.2;','Translates the navigation screen offset horizontally by the specified percent of applet/application window width.','*64','timeSeconds','TRANSLATE','X','x.xx') newCmd('','11.1.1','','*v+11.2;','Translates the navigation screen offset vertically by the specified percent of applet/application window height.','*66','timeSeconds','TRANSLATE','Y','y.yy') newCmd('','11.1.1','','*v+11.2;','Translates the navigation screen offset to the screen position corresponding to the given molecular coordinate.','*68','timeSeconds','TRANSLATE','{{x y z}}','') newCmd('','11.1.1','','*v+11.2;','Translates the navigation screen offset to the screen position corresponding to the geometric center of the specified atoms.','*70','timeSeconds','TRANSLATE','._atom_expression','') newCmd('','11.1.1','','*v+11.2;','Translates the navigation screen offset to the screen position corresponding to the center of the vertices of the specified draw object.','*72','timeSeconds','TRANSLATE','$object','') newCmd('.nbo','13.1.13','','*v+14.4;mo','The NBO command is a variant of the {#.MO~} command and, as such, accepts all of the parameters of that command. It creates isosurface renderings of orbitals created by the {https://nbo6.chem.wisc.edu/nboman.pdf~NBO program}. The following NBO file types are read by Jmol. If any one of the files 32 to 47 are loaded, Jmol will automatically also read file 31 and, if present, file 46, the label file. If file 31 is missing, an error will be thrown. If version 3 of NBO was used (Gaussian plugin, for example), then the 46 file will not be present, and Jmol will look for the output.properties file. If that file is not found, Jmol will simply label the orbitals 1-N. These files are created using the NBO input $NBO PLOT ARCHIVE $END keywords. Note that the "P" version of these orbitals may be used for qualitative visualization. Thus PNAOs "exhibit the idealized spherical symmetries of isolated atoms, but remain optimally adapted in size for the molecular environment. Moreover, these orbitals exhibit the interatomic orbital overlap that underlies qualitative concepts of chemical bonding. PNAOs are therefore the preferred choice as "textbook" atomic orbitals, providing vivid imagery to illustrate the principle of maximum overlap."[ {https://nbo6.chem.wisc.edu/webnbo_css.htm~ref} ] For more information about NBOs, see {https://nbo6.chem.wisc.edu/webnbo_css.htm~What are NBOs} and the {https://nbo6.chem.wisc.edu/nboman.pdf~NBO Manual}.[||31|AO|Atomic Orbital basis functions (required)|| 32| PNAO|Preorthogonal Natural Atomic Orbitals ||33|NAO|Natural Atomic Orbitals || 34| PNHO|Preorthogonal Natural Hybrid Orbitals || 35| NHO|Natural Hybrid Orbitals || 36| PNBO|Preorthogonal Natural Bond Orbitals || 37| NBO|Natural Bond Orbitals || 38| PNLMO |Preorthogonal Natural Localized Molecular Orbitals || 39| NLMO|Natural Localized Molecular Orbitals || 40| MO|Molecular Orbitals || 44| PRNBO |Preorthogonal Resonance Natural Bond Orbitals (NBO 7) || 45| RNBO |Resonance Natural Bond Orbitals (NBO 7) || 46|labels|optional descriptive labels, such as "C1-C2" (NBO 7) || 47 |archive|NBO program archive file; GenNBO input file||]','0|1','','') newCmd('','','','','An orbital type, one of: AO, PNAO, NAO, PNHO, NHO, PNBO, NBO, PNLMO, NLMO, MO, PRNMO, or RNMO. Specifying the orbital type results in immediate loading of the corresponding ""plot"" file xxx.32-xxx.41 if present. This command only functions after an NBO output file with orbitals has been loaded (files 31-45 or file 47). The TYPE keyword can be used together with a number or label: NBO TYPE NHO 3, for example.','*2','TYPE','(orbital type)') newCmd('','','','','Specify an orbital by number, 1-based.','*3','(integer)','') newCmd('','','','','Specify an orbital by NBO label','*5','"label"','') newCmd('','','','','Specify a BETA orbital by number, 1-based.','*4','(integer)','BETA') newCmd('.parallel/process','','','*v+12.0;','Jmol is able to use multiple processors on a multiple-CPU machine. Basically what you can do is to tell Jmol which statements in a script you want to run in parallel, and it will do that. The way this is done is to create a function using the keyword PARALLEL in place of the keyword function. Within that block of code, any group of commands surrounded by PROCESS{{ }} will be collected and run in parallel just before Jmol returns from the function. Any commands NOT within these sets will be run BEFORE any PROCESS commands. For example:[||parallel twoIsosurfaces(model1, model2) {{:>var x = 1:>process {{:> isosurface s1 model @model1 molecular; color isosurface red
}}:>process {{ :> isosurface s2 model @model2 molecular; color isosurface green \n }}
x = 2
}}:>
load files "1crn.pdb" "1blu.pdb"
twoIsosurfaces("1.1", "2.1")
frame *||]In this case, the variable x will be 2 BEFORE the isosurfaces are created. See also {examples-11/data/multi-mo.txt~multi-mo.txt} and {examples-11/data/multi-surface.txt~multi-surface.txt}, and {examples-11/data/multiProcessTest.txt~multiProcessTest.txt}. You can selectively turn on and off the use of multiprocessors using {#.set_multiprocessor~set multiProcessor}. If this setting cannot be set true, then it means you do not have a multiprocessor machine. Not all processes will work; currently the only implemented parallel processes are for isosurfaces and molecular orbitals. The parallel capability of Jmol should be considered experimental at this time.','1|1','','') newCmd('.pause','','','*v+11.8 -- adds stepping capability;pause','Note: PAUSE and RESUME are deprecated. see {#.throw~}. Pauses script execution until {#.resume~}, {#.step~}, {#.quit~}, or {#.exit~} is issued. Any text on the command line after pause is send to the user as a message when the script pauses. During the paused condition, commands can be entered from the Jmol application console, and they will be executed. The next command to be executed can also be checked by issuing ? from the console.','1','message','') newCmd('.[plane expressions]','','','*v+14.32 -- adds HKL OFFSET;iso;atoms','Several commands in Jmol accept parameters that define planes. The commands {#.depth~}, {#.draw~}, {#.isosurface~}, {#.mo~}, and {#.slab~} all have the PLANE option, and {#.depth~}, {#.draw~} {#.isosurface~}, and {#.slab~} also have the HKL option for defining planes using Miller indices. In addition, {#.select~}, {#.restrict~}, {#.display~}, and {#.hide~} as well as many other commands accept {#.atomexpressions~atom expressions}, and these may include one or more WITHIN() function. The method of describing planes in Jmol is defined below:[||hkl|Miller indices are simply indicated by enclosing them in brackets, usually as fractions: {{h k l}}. For example,
isosurface hkl {{0 1/2 1/2}}
. ||plane|A plane is defined in Jmol in one of five ways: (a) listing three points, (b) giving the parametric equation ax + by + cz + d = 0, (c) referencing an already-defined {#.draw~} or {#.isosurface~} object that describes a plane, (d) using the strings "xy", "xz", or "yz", or (e) using a simple single-variable equation such as "x=3" or "y=-2". Points may be embedded atom expressions (that is, atom expressions surrounded by parentheses), for which the center is used, Cartesian coordinates expressed as {{x,y,z}}, or crystallographic fractional coordinates indicated using "/" in at least one coordinate: {{0,1/2,1/2}}. Commas are optional. Any combination of these three point types may be used. For example:
display within(0,plane,@{{plane({{atomno=3}}, {{0 0 0}}, {{0 1/2 1/2}})}})
The parametric equation ax + by + cz + d = 0 is expressed as {{a b c d}}.
Planes based on draw and isosurface objects are first defined with an ID indicated, for example:
draw plane1 (atomno=1) (atomno=2) (atomno=3)
After that, the reference $plane1 can be used anywhere a plane expression is required. For instance,
select within(0,plane, $plane1)
or
isosurface PLANE $plane1
These objects can be defined and then hidden from the user using draw off or isosurface off. For selection purposes the plane is considered of infinite extent even if it only shows up as a small triangle or quadrilateral. ||] ','0','','') newCmd('.plot','12.0.RC23','','*v+12.0 replaces quaternion and ramachandran commands, and adds new property options;plot, draw','Jmol can create a variety of Ramachandran plots and quaternion maps. In addition, Jmol has the capability to quickly generate simple plots relating two or three atom properties. A new frame is created. In the case of property and Ramachandran plots, this frame has its own independent orientation; in the case of quaternion maps, rotation of the map is synchronized with rotation of the model. Only one data frame may be visible at a time. Also related to this command for quaternions and Ramachandran plots is the {#.draw~} command, which allows depicting of these measures on the model itself. ','0|1','','') newCmd('','','','plot','Creates the quaternion representation of the protein or nucleic acid in the three dimensions not given by the specified axis.','*21','QUATERNION','w, x, y, or z') newCmd('','','','plot','Creates the quaternion difference representation of the protein or nucleic acid in "w" projection. "a", for absolute, produces a visualization of q[i] / q[i-1], which defines the helical axis of a helix or beta-pleated sheet; "r", for relative, produces a visualization of q[i-1] \\ q[i], which defines the relative rotation of the (i)th residue from the perspective of the (i-1)th residue, defining the pitch of the helix. The default visualization for "r difference" (utilizing set quaternionFrame "c") for a typical protein is an ellipse having a major axis tilted at 36 degress from the quaternion X axis and minor axis along the quaternion Z axis. This ellipse represents a composite rotation from one amino acid to the next involving a rotation about the q[i] frame Z axis (perpendicular to the N-CA-C plane) of approximately 180 - 110 degrees ((180-110)/2 = 36 degrees), followed by a rotation about the q[i] frame X axis (CA-C bond) of 180 + psi[i-1] + phi[i] degrees.','*22','QUATERNION','a,r','DIFFERENCE','') newCmd('','','','plot','Creates a visualization of dq[i+1] / dq[i], where dq is defined as q[i+1] / q[i] for "a" or q[i] \\ q[i+1] for "r". In the case of "r difference2", for most amino acids this is a point relatively close to the quaternion x axis. ','*23','QUATERNION','a,r','DIFFERENCE2','') newCmd('','','','plot','Creates a Ramachandran plot for a the currently displayed protein model. Values of PHI and PSI are available directly from the points involved using, for example, averageHelixPsi = {{2.1 and helix}}.psi.','*31','RAMACHANDRAN','') newCmd('','','','plot','Creates a "relative" Ramachandran plot for a the currently displayed protein model with the third axis being theta, a measure of "straightness" as calculated using a Ramachandran angle approximation: straightness[i] = 1-acos(abs(cos(theta[i] / 2)))/90, where i refers to a residue and theta is the angle associated with the relative quaternion second derivative dq[i]\\dq[i-1]. Theta can be approximated using Ramachandran angles for "C" straightness simply as theta =approx= psi[i] - psi[i-1] + phi[i+1] - phi[i] (Hanson and Kohler, unpublished results) and for "P" straightness as follows: If deltaPhi = phi[i+1] - phi[i], deltaPsi = psi[i+1] - psi[i], and alpha = 70o, then cos(theta/2) =approx= cos(deltaPsi/2)cos(deltaPhi/2) - sin(alpha)sin(deltaPsi/2)sin(deltaPhi/2). (Hanson and Braun, unpublished results.) Residues close to the phi-psi plane have low values of theta and thus high values of straightness, meaning they are in a region of relatively high structural regularity --- usually helices or sheets.','*32','RAMACHANDRAN','r') newCmd('','','','plot','Creates a 2D plot relating two atom properties for the currently selected atom set. For example: select *;plot properties atomno temperature. Additional optional parameters MIN {{x y z}} and MAX {{x y z}} may follow. Setting these values sets the scale of the graph within Jmol and truncates data values outside of this range. (The z value in this case is ignored.)','*11','PROPERTIES','property1','property2','') newCmd('','','','plot','Creates a 3D plot relating two atom properties for the currently selected atom set. For example: select *.CA;plot properties phi psi resno. Additional optional parameters MIN {{x y z}} and MAX {{x y z}} may follow. Setting these values sets the scale of the graph within Jmol and truncates data values outside of this range.','*12','PROPERTIES','property1','property2','property3') newCmd('.pmesh','','','*v+11.8 merges pmesh and isosurface commands;iso','The pmesh command is deprecated. See {#.isosurface~} for information about the pmesh file format options.','0|1|2','','') newCmd('.polyhedra','','polyhedra1,polyhedra2','*v+11.0 -- expanded capability, new algorithm;iso,poly','Jmol will form a wide variety of polyhedral structures. The polyhedra command can be used for either construction of new polyhedra or modification of existing polyhedra. Starting with Jmol 14.4, you can create polyhedra for crystal structures for which symmetry has not been applied or necessary atoms have not been loaded (the UNITCELL option). All that is necesary is the central atom. In addition, you can get information about a polyhedron using getProperty("shapeInfo.polyhedra[1]").
Finally, you can select polyhedra and use the polyhedra() or polyhedra(n) functions to get the atom centers of all or selected polyhedra.','0|1|2','','') newCmd('','','','','Used for construction of new polyhedra, parameters fall into five subsets:
polyhedra [basis] [selection sets] [display options]. ','TEXT','Polyhedron construction','') newCmd('','','','','Starting with Jmol 14.4, polyhedra in crystal structures can be created where only the central atom may be loaded. (Answering, for example, the question: What is the atom environment around Au3 in this crystal structure?)
load t.cif // no symmetry applied just the unit cell, maybe just one atom
polyhedra 4-12 UNITECELL @1
In addition, Jmol can calculate, show, and draw the point group of a polyhedron:
load t.cif
polyhedron 12 unitcell @1
select @1
show pointgroup POLYHEDRON
draw pointgroup POLYHEDRON
You can also draw points from the derived array using the {#.draw~DRAW POINTS} option along with getProperty():
draw diameter 0.2 POINTS @{{getProperty("shapeInfo.polyhedra[1].vertices")}}
','TEXT','Polyhedra and symmetry','') newCmd('','','','','Polyhedra involve a central atom and 3 or more outer "vertex" atoms. Polyhedra can be formed based either upon the number of atoms bonded to the central atom (polyhedra 4 BONDS) or upon the number of atoms within a specified range of distance from the central atom (polyhedra RADIUS 1.5 2.0), or a combination of these (polyhedra 4,5,6 BONDS RADIUS 1.5 2.0). Multiple possibilities for bonds can be indicated using commas (polyhedra 4,6,8) or with a hyphen (polyhedra 4-20) . If only one number is given with RADIUS, then it is considered a maximum distance. One or the other, number of vertices or radius range, must be indicated. If both BONDS and RADIUS are specified, then polyhedra of the given type(s) are constructed only to the specified set of connected atoms that are within the specified range of radius of the central atom. Finally, the keywords BONDS and RANGE may be omitted provided bonds are indicated as integers and distances are indicated as decimal numbers.','TEXT','[basis]','') newCmd('','','','','Potential polyhedra centers and vertex atoms are specified in the form of a standard Jmol embedded {#.atom expressions~atom expression}, such as {{titanium}} or {{atomno<12 and not nitrogen}} and as such must be specified in parentheses. The first set specifies the centers; the second set specifies the vertex atoms. The optional keyword TO can preceed the vertex set for clarity, as in polyhedra 4,6 BONDS {{titanium}} TO {{oxygen or nitrogen}}. If no atom sets are designated, the assumption is "{{selected}} TO {{*}}". A single designation indicates centers, with "TO {{*}}" implied. If only TO and an atom set is specified, then the centers are taken as the currently selected set of atoms.','TEXT','[selection sets]','') newCmd('','','','','Three sets of display options can be included when constructing polyhedra.
- Polyhedra can be displayed either as traditional "FLAT" faces (the default) or as "COLLAPSED" faces, which display more clearly the central atom. An empirically adjustable parameter, distanceFactor can be set to a higher value to include more faces in a polyhedron if some do not form. Its default value is 1.85. For collapsed polyhedra, the distance in angstroms from the central atom to the point of collapse can be specified using faceCenterOffset=x.x where x.x is a distance in Angstroms.
- Polyhedra can be displayed either with no edges (NOEDGES, the default), or with a thick line on all edges (EDGES), or on just the front edges (FRONTEDGES) or edges only (EDGESONLY). The front-edge-only option is only meaningful when the polyhedra are translucent (see below).
- Polyhedra can be colored by indicating a valid color (e.g. red or yellow) or a hexadecimal RGB color triple (e.g. [xFF0000] or [xFFFF00]). The keywords TRANSLUCENT and OPAQUE can also be used. Alternatively, after polyhedra are created they can be colored using the {#.color (model object)~color polyhedra} command.
To turn on, turn off, or delete selected polyhedra, use polyhedra ON, polyhedra OFF, or polyhedra DELETE, respectively. Any of the display options (FLAT, COLLAPSED, EDGES, FRONTEDGES, or NOEDGES) can also be similarly modified. Colors or translucency, however, cannot be applied this way. To color a set of polyhedra that are already formed or to make them translucent, first select the set of centers of the polyhedra to modify, then use the {#.color (model object)~color polyhedra} command.','TEXT','Polyhedron modification','') newCmd('','','','','Create polyhedra automatically, using the maximum gap methodSee Zur Abgrenzung der Koordinationssphäre und Ermittlung der Koordinationszahl in Kristallstrukturen, G. O. Brunner, D. Schwarzenbach,{https://www.degruyter.com/view/journals/zkri/133/1-6/article-p127.xml~ Zeitschrift fur Kristallographie - Crystalline Materials, 1971, vol 133, issues 1-6 127-133. See also {#.polyhedra~POLYHEDRA -x.x};','*11','AUTO','') newCmd('','14.3.15','','*v+14.4;','Removes all renderings except polyhedra.','*12','ONLY','') newCmd('','','','','Set the maximum gap for POLYHEDRA AUTO to be x.x in Angstroms.','*13','-x.x','') newCmd('','','','','Creates polyhedra based on minimum and maximum distances to vertices.','*14','minDistance','maxDistance') newCmd('','','','','Create all polyhedra at the specified centers to vertices that at in the specified set and also bonded to a specific center.','*2','{{centers}}','{{vertices}}') newCmd('','','','','Create one polyhedron with the specified center to the specified atoms. If more than one atom is in {{center}}, only the first atom will be used.','*3','{{center}}','TO','{{vertices}}','') newCmd('','14.4.4','','*v+14.6;','Adds spheres of the given diameter to the points of a polyhedron, colored based on the atom at that position.','*41','POINTS','x.x') newCmd('','14.24.2','','*v+14.6;unitcell','Generates the nth Brilliouin zone as a polyhedron (default 1); can be scaled by adding a SCALE x.x option: polyhedra scale 3.0 BRILLIOUIN 1','*42','BRILLIOUIN','n') newCmd('','14.24.2','','*v+14.6;unitcell','Generates the Wigner-Seitz unit cell as a polyhedron','*43','WIGNER','') newCmd('','14.24.2','','*v+14.3;','Creates a polyhedron specified by the range of vertex count provided by nrange) around each of the currerntly selected atoms that has a matching bonding environment. Ranges can be single numbers (5), comma-separated options (4,6), or ranges (4-8). Does not require atoms at these positions, as long as the central atom is present, using the unit cell and periodicity to find the relevant atom positions. Will check bonding as necessary using autobonding parameters. Accepts all standard POLYHEDRA parameters. ','*44','nRange ','UNITCELL') newCmd('','14.24.2','','*v+14.3;unitcell','Creates a polyhedron specified by the range of vertex count provided by nrange)around each of the currerntly selected atoms that has a matching bonding environment with a limit to the distance from the central atom to a vertex. ','*45','nRange ','maxDistance','UNITCELL','') newCmd('','11.3.17','','*v+14.6;','Create a named polyhedron with arbitrary center and vertices.
For example:
polyhedra ID "myid" {{0 0 0}} TO [{{1 1 -1}}, {{1 -1 1}}, {{-1 1 1}}, {{-1 -1 -1}}] # tetrahedron around origin
polyhedra ID "myid" @{{ {{selected}}.xyz}} TO @{{ {{selected}}.xyz.all}} # polyhedron to center of selected atoms','*5','ID','"id"','{{x y z}} TO {{vertices}}','') newCmd('','11.3.17','','*v+14.6;','Allows a cartesian offset of the named polyhedron.','*6','ID','"id"','OFFSET','{{x y z}}') newCmd('','11.3.17','','*v+14.6;','For x.x > 0 scales a named polyhedron by the given absolute amount; for x.x < 0, moves the polyhedron from the origin by -x.x times its offset.','*7','ID','"id"','SCALE','x.x') newCmd('','14.9.2','','*v+14.9;','Creates a polyhedron based on a set of faces (an array of array of integers, not necessarily wound correctly) and an (optional) set of points in the form of an atom bitset or an array of points (defaulting to {{*}}). For example:
:>load $p4:> polyhedra ID p4 @{{{{*}}.find("*1**1","map")}} creates a fully faced tetrahedron.','*7','ID','"id"','[faces]','[points]') newCmd('.print','11.3.17','','*v+11.4;','Prints a {#.Jmolmath~Jmol math} expression to the console window, status message callback function, and Jmol.scriptWait return value. By itself, print clears the JavaScript and Jmol consoles.','0|1','[math expression]','') newCmd('.prompt','12.0.RC26','','*v+12.0 -- new;','The prompt command pauses a script until the user presses OK or ESCAPE. See also the {#.functionsx=fyfunctions~prompt()} function.','1|2','','') newCmd('','','','','If no parameter is given, displays a trace of the script stack leading to this command. This may be useful for debugging script files. ','*1','','') newCmd('','','','','Displays the message and waits for the user to press OK.','*2','"message"','') newCmd('.quaternion','','','*v+12.0 (deprecated);plot','This command has been superceded by the {#.plot~plot QUATERNION} command.','0|1','','') newCmd('.quit','','','*v+11.0 -- new;pause;quit','When the {#.set_scriptQueue~} is turned on, each script waits for the previous to complete. When a LOOP command is involved and the script queue is enabled, the only way to interrupt the looping script is with another script. So, to account for this issue, the roles of quit and {#.exit~} have been expanded. Either quit or exit at the very beginning of a script command halts any previous still-running script. Processing then continues with the second command on the line. Anywhere else in the command, quit and exit abort that script.','0','','') newCmd('.ramachandran','','','*v+12.0 (deprecated);plot','This command has been superceded by the {#.plot~plot RAMACHANDRAN} command.','0|1','','') newCmd('.redo','12.2.RC6','','*v+12.2 -- new;','(application only) same as the Redo console button -- reloads the last undone state.','1|2','','') newCmd('.redoMove','12.2.RC6','','*v+12.2 -- new;','same as CTRL-SHIFT-Z/CTRL-Y -- redoes changes to atom position when in modelKit mode.','1|2','','') newCmd('.refresh','','','rreset',' Forces a screen repaint during script execution. Usually unnecessary, this command is useful during selected looping operations or in conjunction with such commands as {#.javascript~} that otherwise might not initiate a screen repaint. Refreshing is automatically carried out after a script completes unless {#set_refreshing~set refreshing FALSE} has been invoked. Commands that automatically invoke refresh include {#.delay~}, {#.move~}, {#.moveto~}, {#.navigate~}, {#.write~}, and {#.zap~}. ','0','','') newCmd('.reset','','','*v+11.2 -- adds variable reset;rreset','Resets all model orientation or the value of a variable','0|1','','') newCmd('','','','*v+11.2;','Resets all models to their original position: zoom 100; center; translate x 0; translate y 0. Spinning is not affected (see RESET SPIN, below). Note that if the {#.invertSelected~}, {#.rotateSelected~}, {#.translateSelected~} commands have been given, these changes are not reset, because these changes are recorded in the underlying coordinate set. If it is desired to save and restore atom positions after moving atoms relative to each other, the following commands may be issued: select *;translateSelected {{0 0 0}};save state;..(move atoms here)...;restore state.','*1','','') newCmd('','11.3.29','','*v+11.2;','Resets all aromatic bonds to the type AROMATIC if they are AROMATICSINGLE or AROMATICDOUBLE. Used in conjunction with {#.connect~} and {#.calculate~calculate aromatic} .','*2','AROMATIC','') newCmd('','','','*v+12.2;','Resets the _errorMessage variable.','*3','ERROR','') newCmd('','11.3.23','','*v+11.4;math','Deletes all user-defined functions.','*41','FUNCTIONS','') newCmd('','14.3.11','','*v+14.4;','Resets lighting parameters to Jmol defaults: set ambientPercent 45; set diffusePercent 84; set specular true; set specularPercent 22; set specularPower 40; set specularExponent 6; set celShading false; set celShadingPower 10; set zShadePower 3; set zDepth 0; set zSlab 50; set zShade false;','*45','LIGHTING','') newCmd('','14.1.16','','*v+14.2;','Clears the print buffer. (Useful when using the JavaScript function Jmol.evaluateVar.)','*50','PRINT','') newCmd('','12.1.10','','*v+12.2;','Does a standard RESET, but also turns off spinning.','*51','SPIN','') newCmd('','','','*v+12.2;','Resets PDB HELIX, SHEET, and TURN designations to their default file settings after, for example, using calculate STRUCTURE DSSP.','*6','STRUCTURE','') newCmd('','12.1.10','','*v+12.2;','Resets the default van der Waals radii to standard Jmol values.','*7','VDW','') newCmd('','','','*v+11.2;math','Resets the variable with the given name to the "unset" state.','*8','variableName','') newCmd('','11.3.23','','*v+11.4;math','Deletes all user-defined variables (same as RESET VARIABLES).','*11','ALL','') newCmd('','11.3.23','','*v+11.4;math','Deletes all user-defined variables (same as RESET ALL).','*9','VARIABLES','') newCmd('.restore','','','*v+11.0 -- NEW;rreset','Restores information saved using the {#.save~} command.','0|1','','') newCmd('','','','*v+11.0;','Restores bonding information changed via the {#.connect~} command; if no save name is given, then the last-saved information is restored.','*1','BONDS','saveName') newCmd('','','','*v+11.0;','Restores a previously saved orientation. If no save name is given, then no time in seconds can be given, and the last-saved orientation is restored immediately. If a save name is given, then the number of seconds over which the restoration should be done may be given as well.','*3','ORIENTATION','saveName','timeSeconds','') newCmd('','','','*v+13.1;','Restores a PyMOL scene with a given name','*4','SCENE','sceneName','timeSeconds','') newCmd('','','','*v+11.0;','Restores a previously saved selection. If no save name is given the last-saved selection is restored. If no saved selection of this name is found, then the action is the same as select none.','*5','SELECTION','saveName') newCmd('','','','*v+11.0;','Retores a previously saved state of the applet. Some of the more complex objects, such as dipoles, isosurfaces, lcaoCartoons, and molecular orbitals are not saved.','*7','STATE','saveName') newCmd('','12.0.18','','*v+12.0;','Retores a previously saved secondary structure. saveName is optional; without it, this command restores the author-designated structure after use of the {#.calculate~calculate structure} command. ','*8','STRUCTURE','saveName') newCmd('','14.1.16','','*v+14.2;','Restores the unit cell to its original setting.','*9','UNITCELL','') newCmd('.restrict','','','dhide','Selects the atoms identified by the {#.atom expressions~atom expression} and deletes all characteristics of all atoms and bonds that are outside the selection set by setting those characteristics "off". For wireframe and backbone, restrict unconditionally follows {#.set_bondmode~set bondmode OR}, ignoring the current bondmode setting, thus removing any bonds or backbone rods involving any atoms not in the restricted set. The restrict command is a holdover from RasMol, kept in Jmol for compatibility only. The {#.display~} command is far more flexible, as it preserves the shape characteristics (size, color, translucency) of the hidden atoms rather than simply deleting the shapes. ','0|1','','') newCmd('','','','','Restricts to all atoms;possibly not H atoms.','*1','{"ALL"}','') newCmd('','','','','Restricts atoms based on an {#.atom expressions~atom expression}.','*2','._atom_expression','') newCmd('','','','*v+12.0;','Restricts atoms based on an {#.atom expressions~atom expression} while respecting the setting of bondmode. ','*3','BONDS','._atom_expression') newCmd('.resume','','','*v+11.0 -- NEW;pause','Note: PAUSE and RESUME are deprecated. see {#.throw~}. Resumes script execution after a {#.pause~}.','1','','') newCmd('.return','','','*v+11.0 -- NEW;flow','Returns from a {#.functions~function} or a script being run with the {#.script~} command. In the case of a function return, may include an optional return value. ','1','returnValue','') newCmd('.ribbon','','structure','disp','Ribbons offer a representation the protein backbone or nucleic acid helix using a flat band. For proteins, control points are chosen to be the center of the peptide bond, and the ribbon is drawn in the direction of the carbonyl oxygen (thus roughly defining the peptide planes). For nucleic acids, the control points are the midpoints between adjacent backbone phosphorus atoms, and the ribbon is drawn in the direction of the C6 carbon. A hermite curve is used.','0|1','','') newCmd('','','','','','*11','._on_off{"ON"}','') newCmd('','','','*v+11.6;','Turns ribbon rendering on and all other rendering (including labels) off.','*12','ONLY','') newCmd('','','','','Normally, ribbons vary in width according to the amino acid atom positions. This command sets the width of the ribbon to be a connstant value (a decimal, in Angstroms). A negative number also implies ONLY.','*2','._ribbon_radius','') newCmd('.rocket','','structure','*v+11.0 -- adds cartoonRocket alternative;disp','Creates a crude "rocket" cartoon. See also {#.cartoon~} in association with {#.set (structure)~set cartoonRockets} for a more precise cartoon with rockets. The {#.set(structure)~set rocketBarrels} option removes the arrow heads from rockets. ','0|1','','') newCmd('','','','','','*11','._on_off{"ON"}','') newCmd('','','','','A negative number also implies ONLY.','*2','._rocket_radius','') newCmd('','','','*v+11.6;','Turns rocket rendering on and all other rendering (including labels) off.','*12','ONLY','') newCmd('.rotate','','quaternions','*v+12.0 -- adds COMPARE, HELIX, SYMOP, TRANSLATE options;rspin;camera','The rotate and spin commands allow single angle rotation or continued rotation (spinning) in one of two specific frames, effectively changing the "camera" position. These frames include the standard "fixed" applet window frame and the internal "molecular" model frame (the axes in the data file). Coordinate specifications for the rotation can be given several ways, including absolute coordinate positions set in braces as {{x, y, z}}, the geometric center of a subset of atoms in the model specified in an atom set in parentheses, for example {{atomno < 10 and not oxygen}}, or the geometric center of a previously defined draw object, indicated by the drawing object name preceded by a dollar sign (for example, $line1). In addition, rotation may be specified to be around six standard axes (x, y, z, -x, -y, and -z) as well as several other options. The keyword MOLECULAR may be necessary for explicit model rotation about the internal molecular axes. Defaults are allowed. The default axis for spinining or rotating is the y (vertical) axis; the default angle of rotation is 10 degrees; the default rotation rate is a slow spin of 10 degrees per second. Options may be given in any order and include: [|| | By itself, the rotate command rotates 10 degrees around the y (vertical) axis in a counterclockwise fashion.|| x, y, z, -x, -y, -z| Rotation about one of the six standard axes. The implied axes are the axes of the window, not the molecule.||x.x | When one number is given, that number indicates for a simple rotation command such as ROTATE x 30 the number of degrees for a rotation, either positive (right-hand rotation) or negative (left-hand rotation). For a spin, for example using ROTATE SPIN y 10, this number indicates the rate of spin in degrees per second. For the TRANSLATION option, this is the number of Angstroms total for the translation.|| x.x y.y | Two numbers, for example, ROTATE 30 10 specifies a finite animated rotation or translation. The first number is as for a single number (above). The second number has an effect that depends upon its sign. If the two numbers are the same sign, as in this example, the second number indicates the rate of spinning in degrees per second (or, for the TRANSLATION option, linear movement in angstroms per second). If the second number is opposite the sign of the first number, as in ROTATE 30 -20 or ROTATE -30 30, then the specified finite rotation or translation is carried out over ¦y.y¦ seconds. In this case, the value of {#.set_waitformoveto~} determines whether the script waits for the rotation or translation to complete or continues while that process occurs independently. || {{atom expression or point}} {{atom expression or point}} | Rotation about an axis pointing from the first point to the second in right-hand direction. For example, rotate {{0 0 0}} {{1 0 0}} 10 is the same as rotate x 10, and spin {{atomno=1}} {{atomno=2}} spins the model around the axis connecting these two atoms. || $drawID | the two atoms desired for rotation can be defined based on a {#.draw~drawn} object such as a line or vector by specifying the ID of that drawn object. || {{3x3 matrix}} | A 3x3 matrix can be used to specify the axis and angle of a rotation; MOLECULAR is implied. || {{4x4 matrix}} | A 4x4 matrix can be used to specify a rotation axis and angle along with a translation; MOLECULAR is implied. || AXISANGLE {{x y z}} | Rotation about an axis defined as a vector from the origin {{0,0,0}} to the specified {{x,y,z}} coordinate. || AXISANGLE {{x y z theta}} | Rotation about an axis through {{0 0 0}} and {{x y z}} by the specified number of degrees. ||HELIX | For any operation involving a translation (4x4 matrix, COMPARE, SYMOP, or TRANSLATE options) carries out the rotation about the optimum helical path. || INTERNAL | Same as MOLECULAR. || MOLECULAR | In the case of simple axes such as x or y, indicates that the axis is defined in terms of molecular coordinates. || QUATERNION {{x y z w}} | Rotation about an axis defined by the specified quaternion in the form of a four-vector. For example, to reset the model to the original orientation without changing the zoom, one can use rotate QUATERNION @{{ !quaternion(script("show rotation")) }}. The keyword QUATERNION is optional.|| SELECTED | Rotate only the currently selected atoms. || SPIN | Provide an animation of the rotation. Implied when two decimals -- a number of degrees and a rate -- are given.|| TRANSLATE | Add a translation to the animation (providing a screw-like motion) MOLECULAR and SELECTED are implied. If only one point is specified, then this command allows for an animation of a movement of the selected set of atoms along the vector connecting {{0 0 0}} and that point. In that case, the extent is in Angstroms and rate is in AngstromsPerSecond; otherwise, as for other rotations, extent is in degrees and rate is in degreesPerSecond. ||]','0|1','','') newCmd('','13.3.4','','*v+14.0;rspin','Rotates the model to a calculated "best" orientation for viewing the selected atoms, such that those atoms\' containing box is thinnest along the line of sight (z), and widest horizontally (x). With ROTATE SELECTED BEST, atom positions are actually changed; otherwise only the viewpoint is changed.','*1','BEST','') newCmd('','13.1.17','','*v+13.2;','A special rotate BRANCH option takes as its parameter a list of n dihedrals with starting and target angles. All dihedrals are rotated concurrently over nSeconds time. The array list has 6n elements, a sequence of for each dihedral of i1, i2, i3, i4, v1, v2, where i1, i2, i3, and i4 are atom indices involved in a dihedral, in order, and v1 and v2 are the initial and final dihedral angles for that set of atoms. See also the {#.compare_bonds~compare BONDS} option, which uses this BRANCH option internally.','*3','BRANCH','@list','nSeconds','') newCmd('','','','','Rotates the atoms that are in the molecular branch containing {{atom 2}} but not {{atom 1}} about the axis connecting these two atoms. For example, to spin a methyl group having carbon atom C12 about its connected atom by 360 degrees at a rate of 30 degrees per second, one might use spin BRANCH {{_C and connected(C12)}} {{C12}} 360 30. If fewer than two numbers are given, rotation will continue until spin OFF or another spin command is issued. Attempting to rotate around a bond within a ring may lead to undesired results.','*2','BRANCH','{{atom1}}','{{atom2}}','') newCmd('','','','','A variation of the {#.compare~compare} command. For atoms, determines the best-fit correlation between atoms in atomSet1 and atoms in atomSet2 and then rotates and/or translates atomSet1 to align with atomSet2. The two atom sets must have the same number of atoms and must have a direct 1:1 correlation of atoms. (For a more general comparison, see the {#.compare~} command. An array of points, such as @{{ {{2.1}}.xyz.all}} may take the place of the second atom set. This allows, for example, saving of a set of coordinates with tempCoords = {{2.1}}.xyz.all and restoring of those coordinates with ROTATE COMPARE {{2.1}} @{{tempCoords}} some time after a selected atom rotation. ','*4','COMPARE','{{atomset}}','{{atomset or coordinates}}','') newCmd('','','','','Carry out the symmetry operation number n (or, if n is a negative number, the reverse of that operation) on the currently selected atoms. ','*5','SYMOP','n') newCmd('.rotateSelected','11.1.13','','*v+11.2 -- new;anim','This command takes all the parameters of {#.rotate~} but only carries out the operation on the currenly selected atoms. In addition, if the keyword SPIN is used, the command starts only the atoms spinning. Note that in its current implementation, this rotate DOES slightly modify atom coordinates; any measurements made after rotating may be off by a fraction of a percent, and continued spinning for a long time may introduced significant errors in relative distances and angles.','0|1','','') newCmd('.save','','','*v+11.0 -- NEW;rreset','Saves a variety of sorts of information for later {#.restore~restoring} either in the current model or a different model.','0|1','','') newCmd('','','','*v+11.0;','Saves bonding information, including atom connections, color, width, and visibility. A save name is optional. ','*1','BONDS','saveName') newCmd('','','','*v+11.0;','Saves a full set of orientation information, including center and position of rotation. A save name is optional but recommended, as it allows restoring of the orientation over a specified number of seconds.','*3','ORIENTATION','saveName') newCmd('','','','*v+11.0;','Saves the current set of selected atoms for restoring later, either for the currently loaded model or for another model. (If used for only the current model, without a load command between save and restore, save/restore SELECTION xxx works the same as {#.define~define xxx selected/select xxx}.) Note that when more than one model are loaded, the set of selected atoms is for the entire set of loaded models, not just the currently displayed one. Applying a restore to a completely different model should be done with care or it may not have the intended results. ','*5','SELECTION','saveName') newCmd('','','','*v+11.0;','Temporarily saves the exact current state of the applet or application in memory for later restoring. Note that from the application or signed applet, using the {#.write~} command, you can save the state to a file on your computer in the form of a Jmol state SPT text file or a state-imbedded PNG or JPG file (which, though appearing as a high-resolution image, can also be "drag-dropped" back into Jmol to restore the state exactly, provided necessary files are in the directory of the image file.) You can also write JMOL files, which are ZIP files that contain all necessary files that have been loaded. ','*7','STATE','saveName') newCmd('','','','*v+12.0;','Temporarily saves the structural state of the model, including definitions of HELIX, SHEET, etc.; can be used before the {#.structure~} or {#.calculate~calculate STRUCTURE} commands to restore the previous structural assignments. ','*8','STRUCTURE','saveName') newCmd('','14.1.11','','*v+14.2;','Saves a pointer to the current point in the script. Same as {#.throw~}, but processing continues, and no error is thrown.','*2','CONTEXT','saveName') newCmd('.script','','','*v+13.0 -- adds arguments to running scripts from spt files;script;javascript','Loads and executes the specified script file/url. The hash/pound/sharp character (#) character marks a comment to the end of the line or a semicolon. The semicolon character (;) separates multiple statements on the same line. A script file may load another script file, up to 10 deep. Within the script, for any files indicated, prepending the file name with $SCRIPT_PATH$ indicates to use the path of script file, not the current path, to find the file. If the command CD $SCRIPT_PATH$ is given in a script, then all file references within that script and any scripts that script references will be relative to the path of that script.
Arguments can be passed to a script using the syntax:
xxx.spt(argument1, argument2,...)
where the script command word is not given, and the arguments are listed in parentheses, like a function. Within the script, these arguments are in the _arguments variable.','0|1','','') newCmd('.script','','','*v+12.0 -- adds LOCALPATH and REMOTEPATH options.;*v-13.0;script;javascript','Loads and executes the specified script file/url. The hash/pound/sharp character (#) character marks a comment to the end of the line or a semicolon. The semicolon character (;) separates multiple statements on the same line. A script file may load another script file, up to 10 deep. Within the script, for any files indicated, prepending the file name with $SCRIPT_PATH$ indicates to use the path of script file, not the current path, to find the file. If the command CD $SCRIPT_PATH$ is given in a script, then all file references within that script and any scripts that script references will be relative to the path of that script.
Arguments can be passed to a script using the syntax:
xxx.spt(argument1, argument2,...)
where the script command word is not given, and the arguments are listed in parentheses, like a function. Within the script, these arguments are in the _arguments variable.','0|1','','') newCmd('','11.9.2','','*v+12.0;','When reading a script created with {#.write~write STATE}, the LOCALPATH keyword instructs Jmol to strip all paths beginning with "file:/" down to the indicated path. So, for example, script LOCALPATH "" "myfile.spt" indicates that all local files referenced in the state script should be read from the current default directory. LOCALPATH can be used with scripts other than state scripts created by Jmol. The mechanism is simply looking for instances of /*file*/"some_file_name". If this construction is found in any script read, the replacement will be made. ','*12','LOCALPATH','"path"','._script_filename','') newCmd('','11.9.2','','*v+12.0;','When reading a script created with {#.write~write STATE}, the REMOTEPATH keyword instructs Jmol to strip all paths beginning with "http:", "https:", or "ftp:" down to the indicated path. So, for example, script REMOTEPATH "data" "myfile.spt" indicates that all remote files referenced in the state script should be read from the subdirectory "data/" within the current default directory. REMOTEPATH can be used with scripts other than state scripts created by Jmol. The mechanism is simply looking for instances of /*file*/"some_file_name". If this construction is found in any script read, the replacement will be made. ','*13','REMOTEPATH','"path"','._script_filename','') newCmd('','','','','Loads and executes the specified script file/url. A hash/pound/sharp character (#) character marks a comment to the end of the line or a semicolon. A semicolon character (;) separates multiple statements on the same line. A script file may load another script file, up to 10 deep. The command word SCRIPT is not necessary if the file is quoted or is of the simple format xxxxx.yyy.','*11','._script_filename','') newCmd('','11.1.9','','*v+11.2;','Just checks the file for proper syntax and the presence of files.','*2','._script_filename','CHECK') newCmd('','11.1.9','','*v+11.2;','Executes command n of the designated script file.','*31','._script_filename','COMMAND','n','') newCmd('','11.1.9','','*v+11.2;','Executes commands n through m of the designated script file. The second number may be ommitted to indicate "to the end of the file": script "myfile.spt" COMMANDS 10 - ','*32','._script_filename','COMMANDS','n - m','') newCmd('','11.1.9','','*v+11.2;','Executes line n of the designated script file. ','*41','._script_filename','LINE','n','') newCmd('','11.1.9','','*v+11.2;','Executes lines n through m of the designated script file. The second number may be ommitted to indicate "to the end of the file": script "myfile.spt" LINES 10 - ','*42','._script_filename','LINES','n - m','') newCmd('','11.3.33','','*v+11.6;','Runs the script command result of the {#.jmolmath~Jmol math expression} in one or more applets. The math expression may be a simple single variable name or quoted string or a more complex expression. If the math expression is a quoted string starting with script such as "script dothis.spt", then the indicated script file will be run in each applet. The applet name can be any of the following.
[|| *|all applets on the same web page as the originating applet (the script will run last in the originating applet)|| >|all OTHER applets|| .(period)|just this applet|| name|the applet named name or, if that does not exist, jmolAppletname. Note that the function getProperty("appletInfo.registry") provides a list of applets on all pages, not just the same page. These other applets may be targeted if their full name (which includes a unique number extension) is given.||"name1,name2,..."|all applets matching the specified names. Note that quotes ARE required in this case.||]
','*5','APPLET','appletName','@{{Jmol math expression}','') newCmd('','11.3.33','','*v+11.4;','Runs the script command result of the {#.jmolmath~Jmol math expression} rather than from a file. The math expression may be a simple single variable name or a more complex expression. For example, var bgColor="red";script INLINE @{{"background " + bgColor}} or var s = "[arg]";script INLINE @{{"select " + s}}.','*6','INLINE','@{{Jmol math expression}') newCmd('','','','','Applet only: Evaluates the return of the indicated JavaScript function as the script to be executed. Execution is blocked by setting Info.allowJavaScript = false for the Info structure used when creating the applet. Note that this is different from the {#.javascript~} command, which simply evaluates the specified JavaScript command. Here the function is evaluated on the embedding page, and the return from that function, which is presumed to be Jmol script, is evaluated within Jmol.','1','javascript:functionCall()','') newCmd('.select','','','*v+12.2 -- adds ADD, REMOVE, and GROUP keywords;dhide','Selects the atoms identified by the expression. If no expression is given then all atoms are selected. The optional parameters ADD and REMOVE prior to the atom expression allow quick alternatives to "selected or..." and "selected and not...", respectively. In addition, the keyword GROUP prior to the atom expression provides a quick alternative to "within(group, ...)". ','0|1','','') newCmd('.select','','','*v+11.6 -- adds extended atom property selection;*v-12.2;dhide','Selects the atoms identified by the expression. If no expression is given then all atoms are selected. A second property expression can be added for more general selection of atoms based on atom properties. ','0|1','','') newCmd('','','','','Selects all atoms (possibly not H atoms).','*1','{"ALL"}','') newCmd('','','','','Selects atoms based on an {#.atom expressions~atom expression}. To select atoms specific to a specific model when more than one model is present, use "/n" where "n" is the model number. For example, to select all atoms of model 3, use select */3. ','*200','._atom_expression','') newCmd('','','','','SELECT and related "legacy" Jmol commands that accept atom expressions, such as DISPLAY, HIDE, and RESTRICT, will recognized Jmol variables as atom expressions if they are bitsets such as ({{0 1 3 5:7}}) or simple integer arrays, such as [0,1,3,5,6,7], or nested numerical arrays, such as [[0,1], [3,5], [6,7]]. But note that these numbers are atomIndex, not atomNo. That is, they start at 0 and increment through all models loaded, never repeating. In addition, these commands will accept variables as the right-hand operand of a comparison operation: x = 33; select atomno=x. In this context, x may also be any appropriate array: x = [1,3,5]; select atomno=x will select up to three atoms in each model loaded.','*201','variable','') newCmd('','14.1.2','','*v+14.2;','Carries out a selection while at the same time turning {#.selectionhalos~} on or off. For example: select on ligand.','*21','ON/OFF','._atom_expression') newCmd('','13.2.1','','','Selects all atoms in any groups containing the specified atoms.','*22','GROUP','._atom_expression') newCmd('','13.2.1','','','Adds the specified atoms to the selected set. May be used with GROUP: select add group within(5,ligand).','*23','ADD','._atom_expression') newCmd('','12.2.1','','','Removes the specified atoms from the selected set. May be used with GROUP: select remove group contact(ligand) and not ligand.','*24','REMOVE','._atom_expression') newCmd('','11.5.40','','','The select command allows for full utilization of Jmol math for atom selection. If the first parameter is an atom expression, such as {{*}} or {{10-30}}, a second parameter can be included. This second parameter must evaluate to a TRUE/FALSE expression involving the individual atoms of the atom expression. Parentheses around the property expression are optional. For example: select {{*.ca}} (atomY < atomX) selects for all alpha carbons for which their X coordinate is less than their Y coordinate. Note that in this context, "x", "y", and "z" are variable names, not coordinates. Use "atomX", "atomY", and "atomZ" if you need to refer to atom coordinates. The variable _x is assigned to the individual atom being tested. This variable can be used in nested select() functions within the select command to represent the atom being tested. For example, select {{*.ca}} (phi < select(y; {{*.ca}}; y.resno = _x.resno + 1).phi)) selects alpha carbons of amino acid residues that have phi angles less than that of the phi angle of the next amino acid in the chain.','*7','._atom_expression','(property expression)') newCmd('','11.?.?','','*v+14.2;','The select BONDS command allows for selection of specific bonds by number (0-based). The selection specifies the exact set of bonds for subsequent COLOR BONDS or WIREFRAME commands. The BONDS parameter can be omitted if the bonds are indicated as a bondset, as in select [{{2 3 5:6}}].','*81','BONDS','({{bond list}})') newCmd('','11.?.?','','*v+14.2;','The select MEASURES command allows for selection of specific already-defined measurement lines. The selection specifies the exact set of measures for subsequentCOLOR MEASURES or MEASURE format commands. For example, select MEASURES ({{0:3}}); color measures blue;measure "2:%1.1VALUE %a1 %a2".','*82','MEASURES','({{measure list}})') newCmd('.selectionHalos','','','*v+11.0 -- NEW;','When ON, Jmol displays {#.halos~} around atoms when they are selected. The radius of the halo is always from 4 to 10 pixels larger than the current setting for {#.spacefill~} or the current setting of halo radius using the {#.halos~} command. The color of any specific halo is determined as described for {#.color selectionHalos~}.','*0','._on_off{"OFF"}','') newCmd('.set','11.1.6','','*v+11.2 greatly expands variable capability;math','Jmol allows a wide range of settings to be changed using the SET command -- see the categories below for details. In all cases in Jmol, "ON" and "TRUE" are equivalent, as are "OFF" and "FALSE". Different words may be used simply because they seem more appropriate for a particular parameter -- to turn an effect on or off; to to enable a feature with true or false. There is really no distinction being made here.
The SET command has a rather convoluted relationship to Jmol math, having been developed long before Jmol math was introduced. In most cases the SET command is no longer necessary -- any simple value you can set with SET can be set simply using an assignment of a value to a variable name:
strandCount = 6
instead of SET strandCount 6. However, it is recommended that you use SET for all {#.jmol parameters~Jmol parameters}, just as a way of clearly indicating in scripts that you are setting a Jmol parameter and not a user variable. Some SET commands accept Jmol math directly: set zDepth mydepth is exactly equivalent to both zDepth = mydepth and set zDepth = @mydepth. Some SET commands do not accept Jmol math directly and instead require the @ notation: set selectionHalos @haloFlag. It is most definitely confusing. A guiding principle is that when using the keyword SET, you can always use @ with one single variable or @{{...}} with an expression. And you can always use actual values; there is no need to use @ with a simple number or string. The table below summarizes the situation:
[||These SET commands have full command replacements and are deprecated. These commands must use @x for variables, because their processing is routed through those specialized commands. Thus, set echo x will not work. For the most part, there is no xxx = counterpart. (The two exceptions being set frank, for which there is showFrank = TRUE/FALSE and set selectionHals, for which there is selectionHalos = ON/OFF.)| set {#.axes~}, set {#.background~}, set {#.boundbox~}, set {#.frank~}, set {#.history~}, set {#.highight~}, set {#.labels~}, set {#.selectionhalos~}, set {#.timeout~}|| The set commands listed on the right require @ with a variable because they have a more complicated context or do not take simple boolean, integer, float, or string values.| {#.set_axescolor~}, {#.set_backgroundmodel~}, {#.set_bondmode~}, {#.set_debug~}, {#.set_defaultcolorscheme~}, {#.set_defaultlattice~}, {#.set_defaults~}, {#.set_defaultvdw~}, {#.set_echo~}, {#.set_energyunits~}, {#.set_fontsize~}, {#.set_formalcharge~}, {#.set_hbond~}, {#.set_historylevel~}, {#.set_measurements~}, {#.set_picking~}, {#.set_pickingstyle~}, {#.set_property~}, {#.set_scriptreportinglevel~}, {#.set_specular~}, {#.set_ssbond~}, {#.set_strands~}, {#.set_structure~}, {#.set_togglelabel~}, {#.set_usercolorscheme~}, {#.set_vanderwaals~}, {#.set_zslab~}||]
','0|1','','') newCmd('','11.3.56','','*v+11.4;','set by itself lists all Jmol parameters that can be set and all Jmol read-only variables (starting with underscore), along with their current values.','*2','','') newCmd('','11.3.56','','*v+11.4;','set followed by a Jmol parameter xxx is the same as set xxx TRUE.','*1','xxx','') newCmd('','','','','set followed by characters ending in question mark lists all Jmol parameters starting with those characters and all Jmol read-only variables starting with underscore and those characters.','*3','xxx?','') newCmd('.set (bond styles)','','setbonds','','This group of commands sets the appearance of various optional bond effects for the model.','2|3','','') newCmd('','','','*v+11.0;bond','In some file formats (.mol files, for example) the connection data may indicate the bond type--single, double, triple, or quadruple. Use set showMultipleBonds OFF when you want all bonds to appear as single bonds. ','*67','showMultipleBonds','ON') newCmd('','','','bond','The default Jmol condition. When script commands affect a set of atoms, BOTH atoms must be in the set for the bonds between them to also be affected. Similarly affects the display of backbone units in the selection of protein residues and nucleic acid bases.','*20','bondMode','AND') newCmd('','','','','When script commands affect a set of atoms, EITHER atom may be in the set for the bonds also to be affected. Similarly affects the display of backbone units in the selection of protein residues and nucleic acid bases.','*21','','OR') newCmd('','','','*v+11.4;bond','Setting this parameter TRUE is equivalent to set bondMode OR; it can be tested using Jmol math.','*22','bondModeOr','FALSE') newCmd('','','','*v+11.4;bond','Sets the global default radius of the displayed bond cylinder in milliAngstroms (default 120). Setting this value to 0 removes all bonds from the display but does not actually "remove" the bonds. Changes to this value are applied immediately to all bonds in all models.','*23','bondRadiusMilliAngstroms','120') newCmd('','','','*v+11.4;bond;hbond','When autobonding, the value of bondTolerance is added to the two bondRadius values for two atoms being tested for a bond. A larger bondTolerance allows atoms that are further apart than the sum of their bondRadius values to still be bonded. This parameter should be adjusted prior to file loading for proper maintaining of the Jmol state. For that reason, it is not recommended that it be adjusted.','*25','bondTolerance','(decimal)') newCmd('','12.0.RC3','','*v+12.0;bond;hbond','For PDB files, generally Jmol will use a hydrogen bond calculation based on RasMol; for other file types, Jmol uses its own calculation. Setting this parameter FALSE causes hydrogen bonds to be calculated for all models using the Jmol calculation and involving {#.hbond~set hbondsAngleMinimum} and {#.hbond~set hbondsDistanceMaximum}.','*52','hbondsRasmol','TRUE') newCmd('','','','*v+11.4;bond;hbond','Setting this parameter TRUE causes hydrogen bonds to be displayed as solid lines rather than dotted lines.','*53','hbondsSolid','FALSE') newCmd('','','','*v+11.4;bond;hbond','Hydrogen bonds between protein amino acid residues or nucleic acid base pairs are displayed as lines. These lines can be displayed whether or not the H atoms are present in the file, and can be drawn either between the two non-hydrogen atoms involved in the bond (O or N, typically, the default) or, alternatively, between the two backbone alpha-carbon atoms, depending upon the desired effect.','*55','hbondsBackbone','FALSE') newCmd('','','','*v+11.0;bond','Sets the minimum bond distance for autobonding. Should be set prior to file loading for proper maintainence of the Jmol state.','*60','minBondDistance','(decimal)') newCmd('','14.3.15','','*v+14.4;bond','Renders multiple bonds as "banana" bonds. This setting must be made each time a new file is loaded.','*62','multipleBondBananas','FALSE') newCmd('','12.1.11','','*v+12.2;bond','Sets the radius of the multiple bond lines relative to the standard setting. Starting with Jmol 14.4, with a positive number for spacing but negative for multipleBondRadiusFactor, the bonds are rotated to be fixed multiple bonds 90 degrees out of the trigonal plane.','*63','multipleBondRadiusFactor','0') newCmd('','12.1.11','','*v+12.2;bond','When changed from its default value of -1, sets the distance between the lines in a multiple bond to a specific value and also instructs Jmol to draw multiple bonds in the plane of the atoms involved. (The default rendering in Jmol is to draw multiple bonds in such a way as they are always visible as multiple bonds.) With a positive number for spacing but negative for multipleBondRadiusFactor, the bonds are rotated to be fixed multiple bonds 90 degrees out of the trigonal plane.','*65','multipleBondSpacing','-1') newCmd('','12.1.46','','*v+12.2;bond','Displays partial bonds as dots, not dashes.','*66','partialDots','FALSE') newCmd('','','','bond','Sulfur-sulfur bonds in cysteine bridges of proteins are displayed as lines. These lines can either be between the two sidechain sulfur atoms (the default) or between the two backbone alpha-carbon atoms, depending upon the desired effect.','*70','ssbonds','BACKBONE or SIDECHAIN') newCmd('','','','*v+11.2;bond','Setting this parameter TRUE is an alternative method of setting disulfide bonds to backbone; it can be tested using Jmol math.','*71','ssBondsBackbone','FALSE') newCmd('','','','bond','Sets the overall scale of all displayed dipole vectors.','*4','dipoleScale','.(-10.0 to 10.0)') newCmd('.set (callback)','11.2adds"jmolscript"inadditiontojust"script"','','*v+11.0 -- NEW mechanism for setting callback functions;javascript','Jmol allows for dynamic JavaScript callback function definition. You can specify the functions to receive callbacks, and you can change the functions at any time. Specific parameters sent to these functions are discussed more fully at {http://jmol.sourceforge.net/jslibrary/#jmolSetCallback~}. To turn off callbacks of a given type, specify "NONE". The function name must be present in JavaScript on the page containing the applet. Specifying "alert" will send the message to the user via a JavaScript alert. Note that quotation marks are required around the function name. If the filename starts with "jmolscript:" then instead of JavaScript being run, Jmol executes the Jmol script that follows. For example, set hoverCallback "jmolscript:script hover.spt" executes the Jmol script "script hover.spt" when an atom is hovered over. The code in the file hover.spt can then respond to that hovering action without the need for JavaScript. ','1|2','','') newCmd('','11.1.45','','*v+11.0;','Sends a message indicating a change of {#.frame~}. For compatibility with Chime, the second parameter of this function returns a number ONE LESS than the actual frame number. This function returns nine parameters. The product of the last two parameters indicates the true direction of animation.
function animFrameCallback(app, frameNo, fileNo, modelNo, firstNo, lastNo, isAnimationRunning, animationDirection, currentDirection)
[||app|String|The name of the applet ||frameNo|int|The current frame number (0-based) ||fileNo|int|The current file number (1-based) ||modelNo|int|The current model number within the current file (1-based) ||firstNo|int|The first frame of the animation range, expressed as fileNo*1000000+modelNo ||lastNo|int|The last frame of the animation range, expressed as fileNo*1000000+modelNo ||isAnimationRunning|int|0 (animation is off) or 1 (animation is on) ||animationDirection|int|1 (animation direction +1) or -1 (animation direction -1) ||currentDirection|int|1 (forward) or -1 (reverse)||]','*1','AnimFrameCallback','"function name"') newCmd('','14.29.24','','*v+14.29;','Sends a message indicating what atoms have been moved.','*3','AtomMovedCallback','"function name"') newCmd('','','','*v+11.0;','Sends a message indicating what atoms is being hovered over, independently of whether {#.hover~} is ON or OFF.','*3','HoverCallback','"function name"') newCmd('','','','*v+11.6;','Sends a message each time the {#.echo~} command is executed. If an EchoCallback function is not defined, these messages go to the MessageCallback function.','*21','EchoCallback','"function name"') newCmd('','','','*v+11.6;javascript','Generally the Jmol applet is allowed to use the eval() function of the host page JavaScript using the {#.javascript~} command (unless execution of JavaScript by the applet has been specifically disallowed by setting Info.allowJavaScript = false for the Info structure used when creating the applet). The setting of evalCallback function must be made prior to jmolAppletCall() using jmolSetCallback("evalCallback","someFunctionName"). It cannot be set using set evalCallback. The callback sends the JavaScript to be evaluated back to the web page for evaluation rather than initiating that evaluation within Jmol. This could be important for the signed applet in order to isolate threads or for debugging applet calls to eval(). It is used in {Jmol Protein Explorer~http://chemapps.stolaf.edu/pe/protexpl}. ','*22','EvalCallback','') newCmd('','','','*v+11.6;','Sends messages indicating the status of script execution. Line-by-line script commands are sent if one has set debugScript TRUE. If a ScriptCallback function is not defined, these messages go to the MessageCallback function.','*94','ScriptCallback','"function name"') newCmd('','','','*v+11.6;','Sends a message indicating the status of measurements made by the user. If a MeasureCallback function is not defined, these messages go to the MessageCallback function.','*5','MeasureCallback','"function name"') newCmd('','','','*v+11.6;','Sends a message that indicates the status of a currently running {#.minimize~minimization}.','*71','MinimizationCallback','"function name"') newCmd('','14.32.2','','*v+14.32;','Sends a message that indicates the status ("ON" or "OFF") of the model kit anytime that status is changed.','*72','ModelkitCallback','"function name"') newCmd('','','','*v+11.0;','Sends a message each time a file is {#.load~loaded}.','*4','LoadStructCallback','"function name"') newCmd('','','','*v+11.0;','Sends a wide variety of messages during script execution.','*6','MessageCallback','"function name"') newCmd('','','','*v+11.0;','Sends a message that depends upon the current status of {#.set picking~}.','*8','PickCallback','"function name"') newCmd('','11.1.29','','*v+11.2;','Sends a message indicating changes of the window size.','*91','ResizeCallback','"function name"') newCmd('','13.3.5','','*v+14.0;','Sends a message indicating that a selection has been made.','*95','SelectCallback','"function name"') newCmd('','14.32.14','','*v+14.32;','Sends a message each time a structure is modified. Parameters include mode, atomIndex and modelIndex, where mode is one of: [|| 1 | assignAtom element/charge change start || -1 | assignAtom element/charge change end || 2 | assignBond start || -2 | assignBond end || 3 | assignAtom position start || -3 | assignAtom position end || 4 | delete atom begin || -4 | delete atom end || 5 | delete model begin || -5 | delete model end ||]','*96','StructureModifiedCallback','"function name"') newCmd('','','','*v+11.6;','The SyncCallback method allows a JavaScript function to intercept and modify or cancel an applet-applet {#.sync~} message. If the called function returns "" or 0, the synchronization is canceled; any other string is substituted for the script and sent to the other currently synchronized applets.','*98','SyncCallback','"function name"') newCmd('.set (debugging)','','','*v+11.0 introduces an adjustable logging level and showScript option;',' ','0|1','','') newCmd('','','','*v+12.0;','Turning this parameter ON sets debugScript ON and logLevel to 5; setting it OFF returns these settings to their standard values (OFF and 4, respectively).','*11','debug','OFF') newCmd('','','','*v+14.15;','Same as set loglevel 6','*12','debugHigh','OFF') newCmd('','','','','Turning this parameter ON enables debugging (going to a JavaScript message {#.set (callback)~callback}).','*2','debugScript','OFF') newCmd('','11.5.4','','*v+11.6;','Sets the maximum delay that scripts will use, primarily used for testing scripts. The default setting of this parameter, 0, disables maximum delay checking.','*31','delayMaximumMs','0') newCmd('','11.7.10','','*v+11.8;','A debugging parameter.','*32','fontCaching','TRUE') newCmd('','','','*v+11.4;','Primarily for debugging complex scripts. Sets the level of depth in scripts for which the command {#.history~} is recorded. A setting of 0 (the default) indicates that commands in scripts run using the {#.script~} command should not be recorded. A setting of 1 indicates that such commands should be recorded for script commands given at the top level. A setting of "n" indicates that all commands executed from script commands through level "n" should be recorded. A setting of -1 turns off all history recording.','*33','historyLevel','(integer)') newCmd('','','','*v+11.0;','You may also set the amount of logging sent to the Java console (as opposed to the Jmol {#.console~}). The default level is 4, "information, warnngs, and errors". Levels include:
[||0|no messages whatsoever||1|fatal errors only||2|all errors||3|all warnings and errors||4|information, warnings, and errors||5|full debugging||]','*4','logLevel','(0 - 5)') newCmd('','14.21.1','','*v+14.21;','Set to TRUE, all time delays in {#.moveto~}, {#.zoomto~}, {#.rotate~}, and {#.spin~} are set to 0, and all file operations are set to be synchronous. Disables {#.delay~} and {#.pause~}.','*50','noDelay','') newCmd('','','','*v+11.0;','Turning this parameter ON causes Jmol to show the script commands from a script file as they are executed. A slight difference may be observed when showScript is set in comparison to normal operation in that when showScript is set, an automatic refresh is executed after every command. ','*51','showScript','OFF') newCmd('','','','*v+11.0;','Shows the script commands from a script file as they are executed and pauses the specified number of milliseconds between commands. Setting the delay to 0 turns script showing off.','*52','showScript','milliseconds') newCmd('','11.1.13','','*v+11.2;','Sets the maximum script depth (how many scripts have been called) for which certain messages should appear in the console and be reported back via callbacks. A value of 0 (default) displays messages only from the topmost level -- as, for example, commands entered from the console; a value of 1 displays messages from the top level and messages due to commands such as "script t.spt" but not from scripts that are called from t.spt; etc. A value of -1 turns off all reporting. Affected commands include {#.connect~}, {#.select~} (and related commands), and {#.set~}. This parameter is particularly useful when scripts utilize {#.goto~message/goto} to control program flow.','*4','scriptReportingLevel','(integer)') newCmd('.set (antialiasing)','11.3.43','','*v+11.4 -- NEW high-resolution rendering;','Antialiasing is the smoothing of jagged lines and sharp boundaries in an image. Jmol independently antialiases the display and image creation (using the {#.write~} or {#.capture~} commands). If the display is antialiased, the option also exists to antialias translucent objects or not. Memory requirements are doubled and rendering performance will be diminished when antialiasDisplay is turned on.','2|2','','') newCmd('','','','*v+11.4;','Turning this parameter ON results in smoothing of the model rendering in the window.','*1','antialiasDisplay','OFF') newCmd('','','','*v+11.4;','Turning this parameter OFF removes smoothing of the translucent components of the window in addition to the opaque components when antialiasDisplay is turrned on.','*2','antialiasTranslucent','ON') newCmd('','','','*v+11.4;','Turning this parameter OFF disables smoothing of the images created using the {#.write~} command.','*3','antialiasImages','ON') newCmd('.set userColorScheme','11.3.13','','*v+11.4 -- NEW customizable user color schemes;color','Sets a custom color scheme. Color values can be names such as "red" or "blue" or hexadecimal numbers as [xRRGGBB] or {{r g b}} triples in the form of 3D points. The scheme can then be referred to as "user"; it\'s reverse as "resu". ','*3','colorName','colorName','...','') newCmd('.set echo','','','*v+11.0 introduces movable echo text and 3D echos, more label control, version 11.6 adds images;label','Sets positioning and visibility parameters associated with text written to the screen using the {#.echo~} command. In all set echo commands, an optional id may be given. If this id is not given, the previously indicated id is assumed. set echo IMAGE allows images to be displayed instead of text. Characteristics of the text that are settable include: [||background color | Includes translucency. See {#.background~background ECHO}. ||depth | The default Z-position for echoed text and images is in front of the model. However, this can be set to any {#.depth~} in relation to the model, from 0 (rear) to 100 (front). See below. ||font color | Includes translucency. See {#.color~color ECHO}. ||font size, face, style, and scaling factor | See {#.font~font ECHO}.||ID "xxx" | The optional ID keyword introduces an echo ID that is in quotes or from evaluation, such as set echo ID @{{"test" + i}} in order to distinguish it from echo text, which would be indicated with set echo "text here". Three special echo IDs, TOP, MIDDLE, and BOTTOM are defined for quick placement of text. These three special echos are always defined along with one of three predefined positions and text alignments: LEFT, CENTER, or RIGHT. Thus we have set echo TOP LEFT, set echo TOP RIGHT, etc. Note that you cannot have two separate echos both with the ID TOP. Thus, after set echo top left;echo OK, if you issue set echo top right, the text "OK" originally in the top left corner will jump to the top right corner. Note that TOP is an echo label, not an echo position. If you need two echos, one on the top left and one on the top right, it is best to use your own label id -- set echo myTopLeft 0 100%; echo "on the left"; set echo myTopRight 100% 100%; echo "on the right". The difference in this case between set echo TOP RIGHT and set echo myTopRight 100% 100% is that in the latter case, if you want to right-align multi-line text, you also need to issue set echo myTopRight right. With set echo TOP RIGHT, the text is both positioned on the right and right-aligned.||IMAGE | You can also place JPEG, PNG, or GIF images at either a 2D screen position or a 3D molecular position. See below. ||model | Echo text can be associated with a specific model, with visibility controlled by the {#.frame~} command. See below. ||scaling | Using the command {#.set misc~set fontScaling TRUE} 3D-positioned images and text along with atom labels can be made to scale when {#.zoom~} is applied. The current zoom setting is used to fix a "scaling factor" used for the font. However the {#.font~} command can be used to set a different reference scaling factor. The absolute scale of an image independent of this 3D zoom scaling, can be set using set echo scale, below.||text alignment| Text in echos is generally left-aligned. You can use set echo myecho CENTER to center text at the given position of that echo or set echo myecho RIGHT to have it right-aligned. Use set echo myecho LEFT to return the text to the default left alignment. Note that the special echo IDs TOP, MIDDLE, and BOTTOM are automatically text-aligned based on their required horizontal postion keyword -- LEFT, RIGHT, or CENTER. If you need center-aligned text that is positioned in the top right corner, for example, use your own ID: set echo myecho 100% 100%;set echo myecho CENTER. Set echo myecho CENTER also aligns multi-line text and images vertically.||visibility | Echos are deleted using set echco off. However, you can selectively display or hide echos using either the set echo HIDDEN/DISPLAYED command or the {#.hide~} and {#.display~} commands with $name, where name is the name given an echo in the set echo command (including the special echos TOP, MIDDLE, and BOTTOM using $top, $middle, and $bottom, respectively). ||x and y position within the window | A "2D" echo. A block of text can be displayed anywhere within the applet or application window. An automatic adjustment is made to prevent text from going outside of the bounds of the window. See below. ||x, y, and z position within the model itself | A 3D echo. This position can be defined as a point in space, the coordinate of an atom, the average coordinate of a set of atoms, the position of a {#.draw~drawn point}, or the average position of a drawn object. See below. ||]
Default settings for font, color, and background can be set by first issuing set echo none, so that no real echo is set, and then issuing the desired {#.font~}, {#.color~}, and {#.background~} commands. In addition, any changes to these settings for any specific echo text become the new default for future text echos that are created for the same model.
','1|2|3','','') newCmd('','','','*v+11.0;','Selects the horizontal text alignment (LEFT, CENTER, or RIGHT) for a user-positioned text echo. For the specific echos with ID TOP, MIDDLE, and BOTTOM, the horizontal alignment is also relative to the window. Any text already displayed in this position is immediately realigned. The CENTER option also centers the text box (or image) vertically at the 2D or 3D point defined for this echo.','*32','ID "id"','._echo_horizontal{left}') newCmd('','','','*v+11.0;','Sets a custom echo position with the given name, where (0,0) is the bottom left corner. The next-issued {#.echo~} command will write text to this position. Any text already displayed is immediately moved. If the text would flow out of the applet window, it is repositioned just inside the boundary.','*33','ID "id"','x-position','y-position','') newCmd('','','','*v+11.0;','Sets echo position based on a percentage of the applet window size. (One or the other or both of x and y can have percent.) Note that set echo myecho 0% 100% is not quite the same as set echo TOP LEFT; set echo myecho 100% 0% is not quite the same as set echo BOTTOM RIGHT. The difference is that TOP LEFT and BOTTOM RIGHT automatically justify multiline text as well. Using percentages allows you to place text anywhere within the window and independently justify the text using set echo myecho LEFT, set echo myecho RIGHT, set echo myecho CENTER.','*340','ID "id"','x%','y%','') newCmd('','','','*v+11.6;','Sets a custom echo position with the given name, where [0 0] is the bottom left corner. The next-issued {#.echo~} command will write text to this position. Any text already displayed is immediately moved. If the text would flow out of the applet window, it is repositioned just inside the boundary.','*341','ID "id"','[x y]') newCmd('','','','*v+11.6;','Sets echo position based on a percentage of the applet window size. Note that set echo myecho [0 100%] is not quite the same as set echo TOP LEFT; set echo myecho [100 0%] is not quite the same as set echo BOTTOM RIGHT. The difference is that TOP LEFT and BOTTOM RIGHT automatically justify multiline text as well. Using percentages allows you to place text anywhere within the window and independently justify the text using set echo myecho LEFT, set echo myecho RIGHT, or set echo myecho CENTER.','*342','ID "id"','[x y %]') newCmd('','','','*v+11.0;','Sets echo position in three dimensions, based on molecular coordinates (which may be fractional). Braces are required.','*35','ID "id"','{{x y z}}') newCmd('','','','*v+11.0;','Sets echo position in three dimensions, based on the geometric center of the specified atoms. Braces are required. "OR" should be used rather than "," in the atom expression unless additional parentheses within the braces are also provided.','*36','ID "id"','{{','._atom_expression','}') newCmd('','','','*v+11.6;','Specifies a Jmol script that will be run when the echo is clicked.','*450','ID "id"','SCRIPT','"script"','') newCmd('','','','*v+11.6;','By default, when multiple models are present, all echo text is visible regardless of the model currently displayable using the {#.frame~} command. The set echo ... MODEL command associates an echo with a specific model, allowing the text or image to appear and disappear when different models are displayed using the {#.frame~} command.','*402','ID "id"','MODEL','(model number)','') newCmd('','','','*v+11.6;','Reads the specified BMP, JPG, GIF, or PNG file and displays that image instead of text. All 2D and 3D options are available. Images can be scaled using set echo scale (below).','*401','ID "id"','IMAGE','"file name"','') newCmd('','14.1.16','','*v+11.6;','Sets the depth of the echoed text or image in percent of the model diameter (0 rear, 100 front). Starting with Jmol 14.2, this setting also applies to echos that have been positioned using {{x, y, z}}. In that case, depth values z >= 1000 are (z - 1000) percent in front of the 3D position, and z <= -1000 are (1000 - z) percent behind the 3D position.','*38','ID "id"','DEPTH','z','') newCmd('','11.6.RC7','','*v+11.6;','Selectively displays an echo that previously has been hidden. The name can be any defined echo, including TOP, MIDDLE, or BOTTOM.','*39','ID "id"','DISPLAYED') newCmd('','','','*v+14.8;','Offsets the label in screen pixels','*420','ID "id"','SCALE','(decimal)','') newCmd('','','','*v+14.8;','Scales an echo image to the desired absolute amount.','*430','ID "id"','SCALE','(decimal)','') newCmd('','','','*v+11.6;','Scales an echo image to the desired absolute amount.','*440','ID "id"','SCALE','(decimal)','') newCmd('','13.1.19','','*v+13.2;','Connects echo text with an atom or point with a line.','*405','ID "id"','POINT','._atom_expression_or_coord','') newCmd('','11.6.RC7','','*v+11.6;','Selectively hides an echo without deleting it. The name can be any defined echo, including TOP, MIDDLE, or BOTTOM.','*42','ID "id"','HIDDEN') newCmd('','','','*v+11.0;','Changes the text of the echo message; same as echo "some text". ','*70','ID "id"','"some text"') newCmd('','','','*v+11.6;','Sets the current echo to be "all echos" for setting font, color, display, hidden, or text of all echos at once.','*43','ALL','') newCmd('','','','','Sets the current echo to be "none" -- The next echo command will be to the console/message callback only. After this command, set echo off will still delete all echos.','*50','NONE','') newCmd('','','','','Turns off (deletes) all echo texts. See also set echo hidden.','*60','OFF','') newCmd('','','','','Sets the overall echo offset on the screen in Angstroms.','*404','ID "id"','OFFSET','{{sx sy sz}}','') newCmd('.set (files and scripts)','','','*v+12.2 -- several new settings, including pdbAddHydrogens;','The following commands relate to how files and scripts are loaded and how scripts are executed. ','0|1','','') newCmd('','11.1.5','','*v+11.2;','When set TRUE (default), Jmol will read scripts that are contained in certain file types and append them to the script set using set defaultLoadScript (below). Embedded scripts are indicated by "jmolscript:" followed by valid Jmol script commands in the following file locations:
[||CIF|any line(s)||MOL|line 3||PDB|any REMARK line(s)||XYZ|line 2||] Support for additional file types will be provided upon user request. ','*10','allowEmbeddedScripts','') newCmd('','11.1.22','','*v+11.2;','Setting this parameter to FALSE causes Jmol to add atoms to the last model in a file set rather than add a whole new model when {#.load~load APPEND} is used.','*11','appendNew','TRUE') newCmd('','','','*v+11.0;','Sets the URL for a proxy server when {#.load~loading} a file or reading a {#.pmesh~} or {#.isosurface~} or when reading a file-based {#.script~}. A proxy server is a server-side application (typically written using PERL, PHP, or ColdFusion) on the same host as the JAR file that can deliver files from other servers on the internet. Jmol appends "?url=" followed by the URL of the requested data file to the indicated proxy server name. NOte that Java security requires that this call be to the same server that hosts the JAR file. ','*12','appletProxy','"URL"') newCmd('','11.1.29','','*v+11.2;bond','Turning this parameter ON instructs Jmol, when applying symmetry to atoms, as in "load xxx.cif {{1 1 1}}", to also apply symmetry to the bonds indicated in the file. The flag is useful when normal Jmol autobonding would not properly connect atoms, but the model is "molecular" -- the base atom coordinates are correct for whole molecules. The flag should not be used in cases where the application of symmetry operations creates new bonds that were not present in the original set, as for quartz.cif, where there is only one bond initially, and after applying symmetry new bonds are created that are between atoms that were created using two different symmetry operations. If bonds are not indicated in a file, this flag is ignored, and Jmol uses its autobonding algorithm to create bonds.','*13','applySymmetryToBonds','OFF') newCmd('','11.7.7','','*v+11.8;load','The MOL2 and MDTOP file formats specifically do not contain enough information within the file to determine the element for all atoms. (The atom types depend upon which force field was employed, and not all force fields allow the direct decoding of element from atom type.) The atomTypes setting allows the user to correlate atom types with specific elements for these specific readers. The syntax is "abcd=>C;efgh=>H;...", indicating that atom type "abcd" is carbon, atom type "efgh" is hydrogen, etc. The matching is case sensitive.','*14','atomTypes','"..."') newCmd('','14.1.11','','*v+14.2;bond','Jmol\'s autobonding algorithm can accept two bonding data sets. The first implemented is from {http://sourceforge.net/p/openbabel/code/485/tree/openbabel/trunk/data/element.txt~Open Babel 1.100.1} and is a mixture of ionic and covalent bonding parameters. set bondingVersion 0 selects this default set. Jmol 14.2 adds a set of covalent bonding parameters from {http://www.ncbi.nlm.nih.gov/pubmed/19058281~Pyykko, P. and Atsumi, M. (2009), Molecular Single-Bond Covalent Radii for Elements 1, Chem. Eur. J., 15: 186}. This second set is purely covalent. In either case, if an atom has a formal charge, or the user as specified the bondingRadius for that atom, neither set will be used.','*15','bondingVersion','0') newCmd('','','','bond','Some file formats that Jmol reads, such as XYZ, do not contain bonding information. In these cases, the default action for Jmol is to generate bonds automatically based on an algorithm. When given prior to loading a model, the set autobond OFF command causes Jmol to not do any automatic bond creation when subsequent models are loaded.','*16','autobond','ON') newCmd('','11.7.30','','*v+11.8;','Specifically for Spartan and Sygress readers, this setting (default FALSE) sets the default orientation (when the file is loaded or {#.reset~} is issued) to that given in the file. Starting with Jmol 12.0, the setting is ignored, and the default orientation is automatically used unless FILTER "NoOrient" is included.','*17','autoLoadOrientation','FALSE') newCmd('','11.6.rc15','','*v+11.6;cd','Sets the path to files specifically for the dialog box that pops up when a file name is "?" in a command such as {#.load~} or {#.write~}. This variable is automatically set when the user navigates to a new directory during a file save dialog.','*18','currentLocalPath','"path"') newCmd('','','','*v+11.0;','Issued prior to the {#.data~} command, sets the text that will separate one set of file datat from another, thus allowing the inline loading of multiple file types.','*19','dataSeparator','"separator text"') newCmd('','','','*v+11.0;cd','Sets the default directory to use for reading all files. This will generally be a relative path such as "./data" or "../files". Note that Java security requires that if the applet is run from a hard drive rather than via the internet, all files read must be either in the directory containing the JAR file or in a subdirectory of that directory. If the applet is unsigned and loaded from the internet, then Java security typically requires that all files read are from the host from which the JAR file was read. But see set appletProxy, above. This flag is applicable only to the Jmol applet, not to the Jmol application.','*20','defaultDirectory','"directory path"') newCmd('','13.1.2','','*v+13.2;','Sets the default script to use for drag-drop and File|Open. The Jmol default is: zap; load ASYNC "%FILE";if (%ALLOWCARTOONS && _loadScript == \'\' && defaultLoadScript == \'\' && _filetype == \'Pdb\') {{if ({{(protein or nucleic)&*/1.1}} && {{*/1.1}}[1].groupindex != {{*/1.1}}[0].groupindex){{select protein or nucleic;cartoons only;}}if ({{visible}}){{color structure}}else{{wireframe -0.1}};if (!{{visible}}){{spacefill 23%}};select *}};','*21','defaultDropScript','"script"') newCmd('','','','*v+11.0;','For crystallographic systems, sets the default lattice to be loaded. {{1 1 1}} loads the standard single unit cell (555). {{2 1 1}} loads two unit cells along the i direction, cells 555 and 655. {{2 2 2}} loads a set of eight unit cells; {{3 3 3}} loads a set of 27. This last is probably most useful, because it loads one unit cell surrounded by all 26 cells sharing its 6 faces, 12 edges, and 8 corners.','*3','defaultLattice','{{i j k}}') newCmd('','','','*v+11.0;','Sets a filter to be the default filter when a file is loaded. For example, set defaultLoadFilter "2D" will automatically convert 2D MOL file data to 3D.','*41','defaultLoadFilter','"format"') newCmd('','','','*v+11.0;','Sets a script to run after any file is loaded. The script must be in quotations. If the script itself needs quotation marks, then it should be placed in a file and indicated as follows: set defaultLoadScript "script myscript.scr".','*41','defaultLoadScript','"script"') newCmd('','','','*v+11.0;','set forceAutoBond ON tells Jmol to disregard any bonding information in a file and use its own internal algorithm for determining connectivity. Its effect is for all future file loads until set OFF. This setting is particularly useful for some PDB and mmCIF files that already have a threshold amount of bonding, so that a full set of bonding can be created automatically at load time. This is necessary for proper assignment of secondary structure. ','*51','forceAutoBond','OFF') newCmd('','','','*v+11.0;','Sets the number of lines of command history to record (minimum 2) and turns history recording ON. set history 0 turns off the command history feature but does not actually set the number of lines to zero. See also {#.history~} and {#.show~show history}.','*55','history','nLines') newCmd('','','','*v+11.0;cd','The load format, by default "http://www.rcsb.org/pdb/files/%FILE.pdb.gz", allows setting of the URL that will be used when "=" is used in front of a file name in a {#.load~} command, for example: load =1crn. ','*61','loadFormat','"URL"') newCmd('','12.1.51','','*v+12.2;','The load format, by default "http://www.rcsb.org/pdb/files/ligand/%FILE.cif", allows setting of the URL that will be used when "==" is used in front of a file name in a {#.load~} command, for example: load ==atp. This parameter also sets the source of ligand files when hydrogens are added using set pdbAddHydrogens TRUE prior to file loading, which allows hydrogen to be added to ligands that are not yet in the PDB component dictionary.','*62','loadLigandFormat','"URL"') newCmd('','12.1.51','','*v+12.2;quit','The load format for reading information such as chemical names, inchikeys, standard inchi, and images from the NIH/NCI CACTVS server, default http://cactus.nci.nih.gov/chemical/structure/%FILE.','*65','nihResolverFormat','"URL"') newCmd('','12.3.29','','*v+12.2;','Sets the path used in all file reading operations such as LOAD, ISOSURFACE, and SCRIPT, regardless of what is given for the path in those commands. Also disables the WRITE command. This parameter can be used to scripts originally pointing to one directory to point to another.','*67','pathForAllFiles','"path"') newCmd('','12.1.51','','*v+12.2;calc','The pdbAddHydrogens option instructs Jmol to add both hydrogens and multiple bonding when PDB files are read. Hydrogen atoms are added to all residues, both ATOM and HETATM. Jmol will fetch RCSB ligand CIF files for attaching hydrogens, and the addition is guaranteed to name hydrogens appropriately. In particular, CH2 groups are named properly, with the proper prochirality. (Not all algorithms do this properly, but Jmol does.) Hydrogen atoms are added to the sidechain nitrogen atom of lysine and to both nitrogen atoms of histidine -- avoiding an analysis of "most probable" hydrogen position. Hydrogen atoms are not added to water. When a state file (SPT JMOL, PNG, or JPG) is created from such a model, Jmol stores the fetched CIF data as part of that state and does not fetch it again when the file is reopened. ','*68','pdbAddHydrogens','FALSE') newCmd('','','','*v+14.21;quit','Turning this parameter OFF disables script queuing. Setting this parameter OFF should never be necessary, but it is provided here as an option. When script queuing is enabled (the default), scripts that are {#.loop~looping} require {#.quit~} or {#.exit~} to be executed in a subsequent script in order to complete. When script queuing is turned off, scripts from different threads may collide and cause unpredictable behavior or crashing of Jmol.','*7','scriptQueue','ON') newCmd('','12.0.RC3','','*v+12.0;load','This parameter sets the maximum number of atoms for default rendering of the model. Models with this number or fewer atoms will be rendered with the default spacefill rendering and the default bond diameter; models with more than this number of atoms will be displayed by default with spacefill 0; wireframe 1 to conserve resources.','*8','smallMoleculeMaxAtoms','40000') newCmd('','12.1.7','','*v+12.2;load','The source of files when using the "$" or "SMILES" keyword in the {#load~} command. Defualt: "http://cactus.nci.nih.gov/chemical/structure/%FILE/file?format=sdf&get3d=True"','*9','smilesURLformat','"URL"') newCmd('.set (highlights)','','','*v+11.0 introduces infinitely variable echos;label','This command group allows for annotation and highlighting of atoms in terms of labels and "halos."','2|3','','') newCmd('','','','disp','(deprecated; see {#.selectionHalos~selectionHalos ON/OFF})','*1','display','SELECTED/NORMAL') newCmd('','','','*v+11.0;','See {#.frank~}','*6','frank','') newCmd('.set (labels)','','','*v+11.0 -- several new customizable features;label','This command group sets parameters associated specifically with atom labels. If the atom expression is not indicated, the action is applied to the currently selected atoms. ','1|2','','') newCmd('','11.5.4','','*v+11.6;','When fontScaling is set ON, any labels created after that are rescaled when the model is {#.zoom~zoomed}. Note that the current zoom setting is taken into account. ','*3','fontScaling','OFF') newCmd('','','','','Sets the font size for atom labels for the currently selected atoms.','*5','fontSize','._fontsize{8}') newCmd('','12.1.50','','*v+12.2;','Sets the default label for PDB atoms. Default: "%m%r"','*1','defaultLabelPDB','"labelFormat"') newCmd('','12.1.50','','*v+12.2;','Sets the default label for non-PDB atoms. Default: "%a"','*2','defaultLabelXYZ','"labelFormat"') newCmd('','','','','Sets the label text alignment within a multi-line label as left-, right-, or center-justified. (For overall label alignment, see set labeloffset, below.) ','*71','labelAlignment','LEFT, RIGHT, or CENTER') newCmd('','','','','Allows setting of label without changing then current selection. Uses same syntax as LABEL command. For example:
:>set labelfor @atoms @myLabel:>set labelfor {{atomno <= 3}} @{{["a","b","c"]}}:>set labelfor {{_C && chirality != ""}} "%[atomname] %[chirality]"
','*72','labelFor','.atom_expression','labelOrArray','') newCmd('','','','','Sets the label offset relative to the atom being labeled. A positive number indicates the number of pixels between the atom center and the beginning of the label. A negative number indicates the number of pixels between the atom center and the end of the label, with the entire label to the left of the atom. Zero in either direction indicates centered for that direction. ','*92','labelOffset','._xoffset','._yoffset','._atom_expression') newCmd('','13.1.15','','*v+13.2;','Sets the label offset relative to the atom being labeled in angstroms in screen space (after perspective rotation). Numbers between 0 and 1 indicate fractions of offset. ','*93','labelOffset','{{x, y, z}}','._atom_expression','') newCmd('','','','','Sets the label offset in PyMOL coordinates, where:
:> sx,sy,sz are screen coord offsets applied after view rotation and sy > 0 LOWERS label:> ax,ay,az are xyz position (in Angstroms; applied before view rotation):>>mode == 0 indicates xyz position is absolute and sx sy sz are Angstroms:>> mode == 1 indicates xyz position is relative to atom position and sx sy sz are Angstroms:>> mode == 2 indicates xyz is absolute, and sx sy sz positions are screen pixels:>> mode == 3 indicates xyz is relative, and sx sy sz positions are screen pixels
','*404','labelOffset','[mode, scrx, scry, scrz, molx, moly, molz]','._atom_expression','') newCmd('','14.3.14','','*v+14.4;','Sets the label offset relative to the atom being labeled. A positive number indicates the number of pixels between the atom center and the beginning of the label. A negative number indicates the number of pixels between the atom center and the end of the label, with the entire label to the left of the atom. ','*94','labelOffsetAbsolute','._xoffset','._yoffset','._atom_expression') newCmd('','','','*v+11.0;','Turning this parameter ON instructs Jmol to add lines pointing from the selected atoms to their respective labels, using the color of the label text ({#.color~color label xxxx}). Note that to see the pointer, the label must be offset from its default (0 0) offset using, for example, set labeloffset 10 10.','*95','labelPointer','OFF','._atom_expression','') newCmd('','','','*v+11.0;','Turns on label pointers to selected atoms, drawing them in the color of the label background.','*96','','BACKGROUND','._atom_expression','') newCmd('','','','*v+11.0;','If a selected label is rotated behind an atom, it is hidden by that atom (default). If an atom expression is given, an indicator of ON or OFF must also be given. OFF is the same as "SET LABELFRONT".','*81','labelAtom','._on_off{"ON"}','._atom_expression','') newCmd('','','','*v+11.0;','The selected labels will always appear in front of all atoms. If an atom expression is given, an indicator of ON or OFF must also be given. OFF is the same as "SET LABELATOM".','*82','labelFront','._on_off{"ON"}','._atom_expression','') newCmd('','','','*v+11.0;','Selected labels appear in an invisible plane just in front of the atoms of their group only. Applicable only to PDB/mmCIF files. If an atom expression is given, an indicator of ON or OFF must also be given. OFF is the same as "SET LABELATOM".','*84','labelGroup','._on_off{"ON"}','._atom_expression','') newCmd('','11.3.52','','*v+11.4;','Toggles the labels on or off for the specified set of atoms. ','*96','labelToggle','._atom_expression') newCmd('','','','*v+14.9;','Specifies whether formal charges are also displayed with {#.label~LABEL} %[nbo].','*97','nboCharge','TRUE') newCmd('.set (language)','11.1.31','','*v+11.2 -- new;','You can set the language for the applet popup menu to a new lanuage just by selecting the desired language from the language submenu.','1|2','','') newCmd('','','','*v+11.2;','The command set language "xx", where xx is a two-letter abbreviation for a lanuage, allows this to be done programmatically. Supported languages are listed on the popup menu.','*1','language','"[two-letter code]"') newCmd('','','','*v+11.8;','Setting this parameter OFF turns off language translation. This may be important for pages that need to parse messages coming from Jmol in American English. ','*2','languageTranslation','ON') newCmd('.set (lighting)','','lighting','*v+11.0 -- clearer naming;disp','This commands in this group determine the overall lighting effects, size, and rotation for the model. Note that in a multi-applet environment, changing lighting values (ambientPercent, diffusePercent, specular, specularExponent, specularPercent, or specularPower) changes them for ALL applets. Effect on another applet may not appear until that model is rotated by the user or some command is issued to that applet that requires updating the display.','2|3','','') newCmd('','','','*v+11.0;','Sets the amount of "ambient" light filling the shadows created by the apparent light source. An ambientPercent value of 0 creates an effect of a spotlight on a stage; a value of 100 removes the shadow entirely, creating a flat, nonrealistic effect.','*10','ambientPercent','(integer 0 to 100)') newCmd('','13.1.13','','*v+13.2;','set celShading TRUE creates a flat effect similar to a hand-drawn figure.','*200','celShading','FALSE') newCmd('','13.3.9','','*v+14.0;','This setting sets the strength of cel shading lines. The default value of 10 is a relatively strong, thick line; 5 is a fine line; 0 turns cel shading off. Negative values remove interior shading, producing only an outline. Pixel based; operates on normal to light source (power > 0) or user (power < 0); sets color to background contrast (black or white) when normal_z < 1 - 2^-(|celShadingPower|/10).','*201','celShadingPower','(integer)') newCmd('','12.1.7','','*v+12.2;','Same as the {#.depth~} command, but note that the parameter can be read and manipulated with math: depth += 10 or if (depth != 0)....','*21','depth','(integer)') newCmd('','','','*v+11.0;','Sets the amount of "diffuse" light apparently emanating from the spotlight, but not hitting and reflecting off the model directly. Setting the diffusePercent value to 0 turns this effect off; giving the effect of the model in a black-walled room where no light reflection is possible, effectively turning off all but specular reflections.','*25','diffusePercent','(integer 0 to 100)') newCmd('','11.9.13','','*v+12.0;','Sets the specular strength using a number which is 2^(specularExponent) ','*3','phongExponent','(integer 0 to 1000)') newCmd('','12.1.7','','*v+12.2;','Same as the {#.slab~} command, but note that the parameter can be read and manipulated with math: slab += 10 or if (slab != 100)....','*50','slab','(integer)') newCmd('','','','','Turns off the specular reflection (the shiny dot on a curved surface).','*51','specular','OFF') newCmd('','','','*v+11.0;','Ranging from 1 to 10, the specular exponent determines the tightness of the specular spot, with 1 being very broad and 10 being very tight. Techincally, log2(phongExponent).','*53','specularExponent','(integer 1 to 10)') newCmd('','','','*v+11.0;','Sets the density of dots in the specular reflection, producing the effect of a very dim light (10) to a very bright light (100).','*57','specularPower','(integer 0 to 100)') newCmd('','','','*v+11.0;','Sets the size of the apparent reflection from a light source.','*55','specularPercent','(integer 0 to 100)') newCmd('','12.1.7','','*v+12.2;','Sets the point where the zShade is fully transparent. Ignored if set equal to zSlab (in which case the settings for slab and depth are used).','*80','zDepth','(integer)') newCmd('','12.1.7','','*v+12.2;','Sets the point where the zShade is fully opaque. Ignored if set equal to zDepth (in which case the settings for slab and depth are used)','*90','zSlab','(integer)') newCmd('','','','*v+11.8;','set zshade ON enables a "fog" or "fade" effect, which shades objects based on the distance from the observer such that distant objects fade into the background. Uses the value of the SLAB and DEPTH settings to determine the point of no effect and full effect, respectively (by default 100 and 0). The settings of SLAB and DEPTH are overridden by ZSLAB and ZDEPTH when ZSLAB is not equal to ZDEPTH. (Both are 0 by default) The effect is enabled regardless of the setting of (slab ON/OFF) and is reset to OFF upon {#.reset~} or the loading of a new model. ','*85','zShade','OFF') newCmd('','','','*v+12.0;','This parameter can be adjusted (typically 1, 2, or 3) to create a stronger effect of fog when zShade is set ON. Setting this value to 0 and then issuing set zshade true displays a black and white shading mask.','*87','zShadePower','(integer)') newCmd('.set (perspective)','','','*v+11.0 -- adds zoomLarge;disp;camera','This commands in this group determine the overall lighting effects, size, and rotation for the model. Note that in a multi-applet environment, changing lighting values (ambientPercent, diffusePercent, specular, specularExponent, specularPercent, or specularPower) changes them for ALL applets. Effect on another applet may not appear until that model is rotated by the user or some command is issued to that applet that requires updating the display.','2|3','','') newCmd('','11.1.1','','*v+11.2;','Sets the amount of perspective. The Jmol default is set cameraDepth 3.0, but you can adjust that to your liking. Smaller numbers lead to more extreme perspective distortion; higher numbers minimize the effect of perspective. The scale in the vertical plane midway from front to back of the model is identical to with set perspectiveDepth off at any zoom level; if p the relative distance of a vertical plane from front (0) to back (1) of the model at a zoom of 100, then the scaling factor is (cameraDepth + 0.5) / (cameraDepth + p) for that plane. This gives, for cameraDepth=3.0, 1.17 for p=0, 1.0 for p=0.5 (as for all cameraDepths), and 0.87 for p=1.','*11','cameraDepth','(positive number)') newCmd('','','','*v+11.2;','Turning this parameter OFF turns off perspective depth. OFF is required for proper function of absolute scale (set scaleAngstromsPerInch nnn). Perspective depth is automatically turned off by Jmol when loading a model having a unit cell and symmetry operators and automatically turned ON otherwise.','*31','perspectiveDepth','ON') newCmd('','','','*v+11.2;','Jmol\'s perspective model involves a linear perspective that is required for navigation mode. Setting this parameter to 10 returns Jmol to the former perspective model used in Jmol 10. When set navigationMode is invoked, the perspective model is automatically set to 11. ','*32','perspectiveModel','11') newCmd('','','','*v+11.2;','Sets the nominal rotation radius for the model effectively setting the size of the model at zoom 100. Normally set to the radius that will contain within the screen the entire model when rotated relative to the default rotation center. ','*40','rotationRadius','(Angstroms)') newCmd('','','','*v+11.2;','Sets the absolute scale of the model by setting the viewing distance from the user to the model in arbitrary units. The actual scale will depend upon the sizes of both the applet window and the screen. ','*43','scaleAngstromsPerInch','._viewing_distance') newCmd('','','','','Setting this parameter to FALSE allows one to set the center of rotation to a new position without also moving that position to the center pixel of the applet window. ','*8','windowCentered','TRUE') newCmd('','','','*v+11.0;','When set FALSE, this parameter disables zooming. Same as {#.zoom~zoom ON/OFF}','*91','zoomEnabled','TRUE') newCmd('','','','*v+13.2;','When this setting is TRUE, Jmol adjusts the zoom only when the height is changed. Adjusting the width only widens the view (as in PyMOL). Setting zoomHeight TRUE disables zoomLarge.','*92','zoomHeight','FALSE') newCmd('','','','*v+11.0;','ZoomLarge is ignored when zoomHeight is TRUE.) When TRUE (Jmol default setting), Jmol adjusts the zoom based on the LARGER of the two application/applet dimensions, height and width. Setting this parameter FALSE causes zoom to be based on the smaller of the two dimensions, which may be useful for the applet if the user is allowed to resize the applet window. PyMOL NOTE: Pymol uses set zoomHeight TRUE, and when a PSE file is read, Jmol automatically goes to this mode.','*93','zoomLarge','TRUE') newCmd('.set (navigation)','11.1.2','','*v+11.2 -- new;nav;disp;camera','This commands in this group set various parameters in relation to navigating through the model. See {misc/navigation.pdf~pdf documentation} for details regarding the Jmol perspective/navigation model.','2|3','','') newCmd('','11.1.3','','*v+11.2;','When navigating, a small square-with-crosshairs pointer usually appears to help guide the user. Setting this parameter TRUE hides that pointer.','*10','hideNavigationPoint','FALSE') newCmd('','11.7.47','','*v+11.8;','Sets the nominal speed of {#.navigate~navigation}. Default value is 10.','*14','navFPS','(integer)') newCmd('','11.1.3','','*v+11.2;','An experimental setting that restricts navigation to the inside of a surface. Not intended for general use.','*1','navigateSurface','FALSE') newCmd('','11.1.3','','*v+11.2;','Sets the depth of the observer navigation point using the same basis as standard slab and depth -- 0 being the back plane of the model; 100 being the front plane of the model. Values greater than 100 represent observation points in front of model.','*2','navigationDepth','(percent)') newCmd('','11.1.2','','*v+11.2;','When set TRUE, enables user-directed navigation through the model using the keypad arrow keys according to the following table. Setting this parameter TRUE automatically sets the perspectiveStyle to 11, sets zoomEnabled, and sets perspectiveDepth on. [||UP/DOWN arrows|go forward/back off||LEFT/RIGHT arrows|turn left/turn right||ALT UP/DOWN arrows|pitch upward or downward||SHIFT LEFT/RIGHT/UP/DOWN arrows|shift navigation point on the screen and in relation to the model||]','*33','navigationMode','FALSE') newCmd('','11.1.5','','*v+11.2;','When set TRUE, and the model has a unit cell defined and symmetry has been applied using {#.load~load} {{i j k}}, creates the effect of an limitless navigation. The navigation center is kept always within the unit cell. The more full unit cells that have been loaded, the more realistic the effect. ','*35','navigationPeriodic','FALSE') newCmd('','11.1.5','','*v+11.2;','Sets the rate of forward/reverse navigation when using the up/down arrow keys. The default value is 5.','*37','navigationSpeed','(integer)') newCmd('','11.1.5','','*v+11.2;','Sets the depth of the depth of the slab plane relative to the observer navigation point using the same basis as standard slab and depth -- 0 (default) being at the navigation point; positive numbers being toward the user relative to that. ','*4','navigationSlab','(percent)') newCmd('','11.7.47','','*v+11.8;','Sets the navigation turning rate in the X direction (yaw) in units of 1/50 degrees per frame.','*51','navX','(decimal)') newCmd('','11.7.47','','*v+11.8;','Sets the navigation turning rate in the Y direction (pitch) in units of 1/50 degrees per frame.','*52','navY','(decimal)') newCmd('','11.7.47','','*v+11.8;','Sets the navigation rate in the Z direction (forward motion) relative to its standard value of 1.','*53','navZ','(decimal)') newCmd('','11.1.3','','*v+11.2;','When set TRUE, the navigation cross-hair cursor is shown whenever in navigation mode, not just while the user is pressing keypad keys or scripted navigation is occurring.','*5','showNavigationPointAlways','FALSE') newCmd('','11.1.2','','*v+11.2;','The visual range is the MINIMUM distance in Angstroms that is viewable by the observer. The default value of 5.0 indicates that with set navigationMode TRUE any object in a plane that is less than 5.0 Angstroms across will be clipped automatically under the presumption that that object is behind the observer. This prevents large objects close to the user from eclipsing smaller objects further from the observer so as to give the illusion of having entered the model itself and now being within it.','*6','visualRange','(angstroms)') newCmd('.set (structure)','','','*v+11.0 -- several new customizable features;','This command group allows for customization of the rendering of PDB and mmCIF secondary structures. The default is set cartoonRockets OFF; set ribbonAspectRatio 16; set hermiteLevel 0; set ribbonBorder 0; set sheetSmoothing 1; set strands 5; set traceAlpha ON.','2|3','','') newCmd('','14.11.2','','*v+14.11;','Setting this parameter ON produces cartoons that have connectors for bases that look like those found in RNA step diagrams (requires load =xxxx/dssr).','*407','cartoonSteps','OFF') newCmd('','13.1.7','','*v+13.2;','Setting this parameter ON produces cartoons that are not just flat ribbons.','*403','cartoonFancy','OFF') newCmd('','13.1.16','','*v+13.2;','Setting this parameter ON enables a PyMOL-like "cartoon_ladder_mode" rendering of nucleic acids, involving a simple stick representation for each base.','*404','cartoonLadders','OFF') newCmd('','','','*v+12.0;','Setting this parameter ON displays nucleic acid bases as triangles that highlight the sugar edge (red), Watson-Crick edge (green), and Hoogsteen edge (blue). See Nasalean L, Strombaugh J, Zirbel CL, and Leontis NB in {http://books.google.com/books?hl=en&lr=&id=se5JVEqO11AC&oi=fnd&pg=PR11&dq=Non-Protein+Coding+RNAs&ots=3uTkn7m3DA&sig=6LzQREmSdSoZ6yNrQ15zjYREFNE#v=onepage&q&f=false~Non-Protein Coding RNAs}, Nils G. Walter, Sarah A. Woodson, Robert T. Batey, Eds., Chapter 1, p 6.','*400','cartoonBaseEdges','OFF') newCmd('','','','*v+12.0;','Displays the nucleotides of RNA and DNA as blocks corresponding to the DSSR convention.','*401','cartoonBlocks','OFF') newCmd('','','','*v+12.0;','Sets the thickness of the blocks representing the nucleosides when cartoonBlocks is set TRUE.','*402','cartoonBlockHeight','0.5') newCmd('','14.1.8','','*v+14.2;','When ON, adds ribose rings to nucleic acid cartoons.','*405','cartoonRibose','OFF') newCmd('','','','*v+11.0;','Setting this parameter ON sets the display of RasMol {#.cartoon~cartoons} to be a mixture of three-dimensional sheet cartoons and alpha helix {#.rocket~rockets}. It is not a precise as a cartoon with respect to helices but more precise than rockets in relation to beta-pleated sheets.','*406','cartoonRockets','OFF') newCmd('','','','*v+12.0;','Setting this parameter FALSE tells Jmol to not use DSSP for the calculation of secondary structure either upon loading a structure with no HELIX or SHEET information or when using the command {#.calculate~calculate STRUCTURE}.','*45','defaultStructureDSSP','TRUE') newCmd('','','','*v+12.0;','Standard DSSP secondary structure analysis disregards all backbone amide hydrogen atoms that might be in a file and instead calculates its own based on the direction of the C=O vector. Setting this parameter FALSE instructs Jmol to not calculate these file-based positions and to instead use the positions of the hydrogen atoms that are in the file. ','*46','dsspCalculateHydrogenAlways','TRUE') newCmd('','','','*v+11.0;','Determines the amount of curve smoothing used in rendering protein and nucleic acid secondary structures involving {#.cartoon~}, {#.ribbbon~}, {#.rocket~}, and {#.trace~}. set hermiteLevel 0 uses a Jmol 10.2 method of rendering these structures, with rope-like traces and paper-thin ribbons and cartoons. Positive values produce more rounded but slower to render shapes, but only when the model is not in motion. Negative numbers produce the same, but also while rotating. A value of 4 or -4 might be a reasonable compromise between look and performance. Hermite levels 6 or higher (-6 or lower) produce a ribbon cross-section in the shape of an ellipse. Use {#.set_ribbonaspectratio~set ribbonAspectRatio 4} rather than the default value of 16 for a more rounded ellipse.','*50','hermiteLevel','(integer, -8 to 8)') newCmd('','11.3?','','*v+11.4;','When set ON, and hermiteLevel is set, draws high-resolution hermite curves even if the diameter is small. Otherwise, small-diameter traces are shown in a faster-rendering fashion.','*53','highResolution','OFF') newCmd('','','','*v+11.0;','Sets the thickness of the ribbons in ribbon and cartoon renderings in terms of the width:height aspect ratio; only enabled in conjunction with set hermiteLevel to a non-zero value. set ribbonAspectRatio 0 turns off this feature; 8-16 is recommended; higher positive numbers leading to thinner ribbons.','*67','ribbonAspectRatio','(integer)') newCmd('','','','','Turning this parameter ON adds a slight border for ribbons.','*68','ribbonBorder','OFF') newCmd('','11.3.38','','*v+11.4;','Turning this parameter ON removes the arrow heads from {#.rockets~} and {#.cartoon~cartoon rockets}, turning them into simple cylindrical barrels.','*70','rocketBarrels','OFF') newCmd('','','','*v+11.0;','In conjunction with set traceAlpha, this parameter determines the "waviness" of beta-pleated sheets. set sheetSmoothing 0 creates wavy sheets, with the ribbon or trace going directly through the alpha carbons. The default is set sheetSmoothing 1, which produces a more averaged, smoother (standard) ripple. Has no effect unless set traceAlpha.','*72','sheetSmoothing ','(0 to 1)') newCmd('','','','*v+11.2;','Sets the number of strands to display for the {#.strand~} and {#.meshribbon~} shapes both, with a maximum of 20.','*91','strandCount','._strand_count') newCmd('','11.3.52','','*v+11.4;','Sets the number of strands to display for the {#.strand~} shape, with a maximum of 20.','*94','strandCountForStrands','._strand_count') newCmd('','11.3.52','','*v+11.4;','Sets the number of strands to display for the {#.meshribbon~} shape, with a maximum of 20.','*92','strandCountForMeshRibbon','._strand_count') newCmd('','','','*v+11.0;','When set TRUE (the default), along with set sheetSmoothing 0, directs Jmol to draw {#.trace~traces} directly through all alpha-carbons. The effect is a wider, more standard alpha helix and a wavier beta-pleated sheet than in Jmol 10.2. Setting set sheetSmoothing 1;set traceAlpha ON directs Jmol to create smooth out the beta-pleated sheets but still follow the alpha carbons for other structure types. With set traceAlpha FALSE, Jmol draws traces through the midpoints of the lines connecting adjacent alpha-carbons, producing tighter alpha helices and smoothed beta-pleated sheets. Also used as the basis for drawing cartoons, meshRibbons, ribbons, rockets, and strands. Jmol has the default settings: set traceAlpha TRUE; set sheetSmoothing 1;set hermiteLevel 0.','*99','traceAlpha','TRUE') newCmd('','12.1.13','','*v+12.0;','Ideally, PDB files will contain header information relating to helix, sheet, or turn locations. When this information is not present, or when the {#.calculate~calculate structure} command is given, Jmol uses an admittedly crude Ramachandran angle range check to determine structure type. The set STRUCTURE command allows setting those Ramachandran angle ranges for helix, sheet, and turn. [phi-psi range] is a set of angles in groups of four. Each group of four numbers includes the minimum and maximum phi values followed by the minimum and maximum psi values that would be valid for this structure type. For example, set structure HELIX [-160, 0, -100, 45] sets "helix" to be defined as phi >= -160 and phi <= 0 and psi >= -100 and psi <= 45 (the Jmol default). The default setting for sheet (really just a strand, since we are not requiring cross-strand hydrogen bonding) and turn are set SHEET [-180, -10, 70, 180, -180, -45, -180, -130, 140, 180, 90, 180]; set TURN [30, 90, -15, 95]. It is recommended that this command be followed immediately with calculate structure;cartoons only.','*96','structure','HELIX|SHEET|TURN','[phi-psi ranges]','') newCmd('.set (visibility)','','','*v+11.0 -- several new features;disp;set','This command group turns on or off specific sets of atoms and axes/cell-related options.','2|3','','') newCmd('','','','*v+11.0;','See {#.axes~}.','*1','axes','._axes_type') newCmd('','','','*v+14.6;','For axes unitcell offsets the axes by the specified fractional coordinants in a, b, and c. Same as axes offset x.x','*201','axesOffset','(decimal)') newCmd('','','','*v+11.4;','Allows setting and checking of the current axes mode: (0) window, (1) molecular, (2) unitcell ','*200','axesMode','0, 1, or 2') newCmd('','','','*v+11.0;','See {#.axes~}; this parameter can be set and checked using Jmol math.','*21','axesMolecular','OFF') newCmd('','11.1','','*v+11.2;','Allows setting of the axes scale; (default 2)','*22','axesScale','(decimal)') newCmd('','','','*v+11.0;','See {#.axes~}; this parameter can be set and checked using Jmol math.','*23','axesUnitcell','OFF') newCmd('','','','','See {#.axes~}; this parameter can be set and checked using Jmol math.','*24','axesWindow','ON') newCmd('','11.1','','*v+11.2;','Sets the background color to the specified value; can be inspected using Jmol math.','*25','backgroundColor','._colorRGB') newCmd('','','','*v+11.0;','See {#.boundbox~}.','*30','boundbox','._axes_type') newCmd('','','','*v+11.2;','This parameter sets the default boundbox color and also can be read as the current boundbox color.','*32','boundboxColor','"color_name"') newCmd('','11.3','','*v+11.4;','Sets the default translucent level (0.5 is standard). ','*33','defaultTranslucent','(decimal)') newCmd('','','','*v+11.0;','set disablePopupMenu TRUE disables the pop-up menu.','*42','disablePopupMenu','FALSE') newCmd('','','','*v+11.0;','Turning this parameter FALSE turns off the unit cell parameter list that is included with {#.unitcell~unitcell ON}.','*44','displayCellParameters','TRUE') newCmd('','','','*v+11.0;','Setting this parameter ON displays all colors as grey scale values.','*46','greyScaleRendering','OFF') newCmd('','','','*v+14.8;boundbox;unitcell','Setting this parameter TRUE displays {#.unitcell~unit cell} and {#.boundingbox~bounding box} edges that are "hidden" to dashed lines, making a more easily visualized cage.','*50','hiddenLinesDashed','FALSE') newCmd('','','','*v+11.0;','Setting this parameter TRUE hides the file name and contents in the pop-up menu starting with the NEXT FILE LOADED.','*51','hideNameInPopUp','FALSE') newCmd('','','','*v+11.0;','Setting this parameter TRUE tells Jmol to hide any atoms whenever they are not selected. set hideNotSelected TRUE immediately hides the currently unselected atoms. When turned off, no immediate action is taken, but future selections no longer hide atoms. The flag is automatically cleared when a new model is loaded or when the picking mode is set to any sort of selection (atom, group, etc.). ','*52','hideNotSelected','FALSE') newCmd('','13.3.4','','*v+14.0;','This parameter replaces {#.set_wireframerotation~set wireframeRotation} and determines how many compromises are used in rendering a model when it is in motion due to animation or user manipulation. The goal is to provide "slimmed down" versions of of the model in order to allow smooth rotation. Larger values include all of the capabilities enabled by lower values. [|| value >= | enables || 8 |antialiasDisplay (and thus, all features)|| 7 |translucency|| 6 |meshes (contact, draw, isosurface, MO, pmesh, lcaocartoon, CGO)|| 5 |cartoons, rockets, trace, ribbon|| 4 |geosurfaces|| 3 |ellipsoids|| 2 |wireframe and balls|| 1 |none of the above (same as "set wireframeRotation off")|| 0 |reserved for "auto"||]','*54','platformSpeed','1 - 8') newCmd('','11.0','','*v+11.0;','When refreshing is set FALSE, no writing to the screen is done at all. Setting refreshing to TRUE should only be done in cases where there is a desire to not show intermediate results as a valid well-tested script runs. If the script throws an error, and refreshing is FALSE, Jmol may appear to be frozen when in fact it is not.','*60','refreshing','TRUE') newCmd('','','','*v+11.0;','Sets the radius of the solvent "ball" that would run around the structure defining its outline. After set radius, you must (re)issue {#.dots~dots ON} for it to take effect, and the solvent probe option for dots must be set on using set solvent ON (below).','*82','solventProbeRadius','._probe_radius{1.2}') newCmd('','13.1.19','','*v+13.2;nmr','Sets the chemical shift reference in PPM for converting magnetic susceptibility to chemical shift. XX is a nucleus such as H or C. For example, set shift_H 30.','*67','shift_XX','(decimal)') newCmd('','','','*v+11.0;','Setting showAxes TRUE displays the axes at their default line width; setting it false hides the axes.','*68','showAxes','FALSE') newCmd('','','','*v+11.0;','Setting showBoundBox TRUE displays the bounding box as a thin dotted line; setting it false hides the bounding box.','*69','showBoundBox','FALSE') newCmd('','','','*v+11.0;','Setting showFrank TRUE displays the Jmol frank in the lower right-hand corner. Checking this parameter allows checking to see if the frank is on.','*70','showFrank','TRUE') newCmd('','','','*v+11.0;','When both this parameter is ON and {#.selectionHalos~} are ON, Jmol will display selection halos even if atoms are hidden. ','*71','showHiddenSelectionHalos','FALSE') newCmd('','','','*v+11.0;','Turns on and off display of hydrogen atoms.','*72','showHydrogens','TRUE') newCmd('','','','*v+11.0;','deprecated -- see {#.selectionHalos~}','*73','showSelections','FALSE') newCmd('','14.1.16','','*v+14.2;','Display full unit cell information in the upper left corner when set displayCellParameters and set showUnitCell are both TRUE. ','*761','showUnitcellDetails','TRUE') newCmd('','11.0','','*v+11.0;','Setting this parameter TRUE is equivalent to {#.unitcell~unitcell ON}; can be tested using Jmol math. ','*760','showUnitcell','FALSE') newCmd('','11.9.19','','*v+12.0;','Setting this flag TRUE removes whole atoms and bonds when they are partially clipped. ','*771','slabByAtom','FALSE') newCmd('','11.9.19','','*v+12.0;','Setting this flag TRUE removes whole molecules when they are partially clipped. ','*772','slabByMolecule','FALSE') newCmd('','11.0','','*v+11.0;','Setting this parameter TRUE is equivalent to {#.slab~slab ON}; can be tested using Jmol math. ','*78','slabEnabled','FALSE') newCmd('','','setsolvent','*v+11.0;','Turning this parameter ON turns on a solvent "probe" that can be displayed using dots. After set solventProbe ON, a subsequent {#.dots~dots ON} shows the surface of the aggregate of selected atoms using dots. This surface is defined by the contact of a spherical probe (representing a solvent molecule) rolled over the surface of the selected atoms. The radius of the probe sphere of 1.4 Angstroms approximates a water molecule. The default radius for Jmol is 1.2 Angstroms, which can be changed using set solventProbeRadius. Note that no change in display occurs after set solventProbe ON until the next {#.dots~dots ON} command is encountered. Does not effect the function of {#.isosurface~isosurface solvent}, which draws a surface regardless of the setting of this flag. For a detailed discussion of molecular surfaces, see {http://www.netsci.org/Science/Compchem/feature14.html~}.','*80','solventProbe','OFF') newCmd('','','','*v+13.2;color','Jmol by default considers translucent objects truly translucent. When set FALSE, translucent obects are not translucent to each other, creating an unreal but possibly desired effect similar to the default PyMOL setting transparency_mode 1.','*85','translucent','TRUE') newCmd('','','','*v+11.0;','See {#.unitcell~}.','*90','unitcell','') newCmd('','','','*v+11.2;','This parameter sets the default unit cell color and also can be read as the current unit cell color.','*93','unitCellColor','"color_name"') newCmd('.set (window)','','','','Allows setting of the window dimensions.','0|1','','') newCmd('','','','*v+11.0;','Sets the window width and height.','*20','width','height') newCmd('','','','*v+11.0;','Sets the window width and height from an array.','*21','[width, height]','') newCmd('','','','*v+11.0;','Sets the window dimensions to match the size of an image.','*22','"xxx.png"','') newCmd('.set (measure)','','','*v+11.0 -- several new features;measure','Sets characteristics of the measurement labels and lines. See also {#.measure~}.','0|1','','') newCmd('','','','*v+11.0;','Sets the format of the labels for {#.measure~distance measurements}.','*20','defaultDistanceLabel','"format"') newCmd('','','','*v+11.0;','Same as for defaultDistanceLabel, but for angles. See {#.measure~} for details.','*21','defaultAngleLabel','"format"') newCmd('','','','*v+11.0;','Same as for defaultDistanceLabel, but for torsions. See {#.measure~} for details.','*22','defaultTorsionLabel','"format"') newCmd('','','','*v+11.0;','Turning this parameter TRUE right-justifies measurement labels','*40','justifyMeasurements','FALSE') newCmd('','','','*v+11.0;','Turning this parameter OFF turns off measurement LABELS only, leaving the lines. (To turn off both, use set showMeasurements OFF.)','*63','measurementLabels','ON') newCmd('','','','*v+11.0;','Setting this parameter FALSE turns off measurement lines and labels BOTH. (To turn off just the label, use set measurementLabels OFF.)','*77','showMeasurements','TRUE') newCmd('','','','*v+14.30 -- adds HZ;','Sets the units for measurement display to be one of ANGSTROMS, AU (or BOHR), NM (or NANOMETERS), (PM or PICOMETERS), VDW, HZ, and NOE_HZ. The VDW option displays distances as percent of the combined van der Waals radii of two measured atoms. Thus, a measurement less than 100% indicates a "clash." The HZ and NOE_HZ options display calculated 1H-1H three-bond (dihedral) and 1H-1H two-bond (geminal) J-couplings using methods of Altona or Karplus, depending upon the nature of the dihedral. In addition, using NOE_HZ, if two hydrogen atoms that are more than three bonds apart are the measurement, then Jmol will perform an NOE calculation and use that result for the measurement (only very preliminarily implemented; not validated).','*63','measurementUnits','(distance unit)') newCmd('','','','','Sets the width of the measurement line in angstroms.','*2','measurements','._width_angstroms') newCmd('','','','','Sets the width of the measurement line in pixels.','*3','','._width_pixels') newCmd('','','','','Sets the measurement line to be dotted.','*4','','DOTTED') newCmd('.set (misc)','',' ','*v+13.0 --adds set energyUnits, forceField, isosurfaceKey, meshScale, nmrUrlFormat, vectorSymmetry;anim;misc','In all cases below, "ON" and "TRUE" are equivalent, and "OFF" and "FALSE" are equivalent.','0|1','','') newCmd('','','','*v+12.0;','This parameter is primarily intended for a touch-screen context. Setting this parameter TRUE allows single-point gestures to be detected. Currently the only single-point gesture supported is a "swipe" or "flick" created by a motion of the mouse or finger that is continuing along a line at the time the mouse or finger is released. ','*03','allowGestures','FALSE') newCmd('','','','*v+11.8;','Set this parameter TRUE to allow key strokes in the applet or application window to be interpreted as script commands. These keystrokes will be displayed if the {#.set_showKeystrokes~showKeystrokes} setting is TRUE.','*05','allowKeystrokes','FALSE') newCmd('','12.0.RC15','','*v+12.0;','Set this parameter FALSE to disable {#.set modelkitmode~modelKitMode}.','*071','allowModelKit','TRUE') newCmd('','12.1.21','','*v+12.2;','This setting is used in association with {#.setpicking~set picking dragSelected}, {#.setpicking~set picking dragMinimize}, {#.set_allowrotateselected~set allowRotateSelected}, and {#.set_dragselected~set dragSelected}. The default FALSE value ensures that whole molecules are shifted or rotated rather than individual atoms. Set this parameter TRUE to allow the moving of just the selected atoms (not whole molecules). ','*075','allowMoveAtoms','FALSE') newCmd('','','','*v+12.0;','Set this parameter FALSE to disable multi-touch gestures (two-finger spread for zoom, two-finger drag for translation) within a multi-touch environment.','*08','allowMultiTouch','TRUE') newCmd('','','','*v+11.2;','This setting is used in association with {#.setpicking~set picking dragSelected}, {#.setpicking~set picking dragMinimize}, {#.set_allowmoveatoms~set allowMoveAtoms}, and {#.set_dragselected~set dragSelected}. When set TRUE, it allows user rotation of selected atoms using the mouse; when FALSE, rotation is disabled.','*09','allowRotateSelected','FALSE') newCmd('','','','*v+11.2;anim;misc;image','Same as {#.animation~animation FPS} -- sets the animation delay in frames per second; can be tested, for example, with "if (animationFPS > 10)..."','*100','animationFps','(integer)') newCmd('','13.3.6','','*v+14.0;anim;misc;image','Same as {#.animation~animation MODE} -- sets the animation replay mode; must be in quotes: "ONCE", "LOOP", or "PALINDROME"; can be tested, for example, with "if (animationMode == \'ONCE\')..."','*100','animationMode','"ONCE" or "LOOP" or "PALINDROME"') newCmd('','11.6.RC13','','*v+11.6;','Setting this parameter FALSE disallows picking of atoms. See also set bondPicking and set drawPicking.','*150','atomPicking','TRUE') newCmd('','11.1.22','','*v+11.2;','Setting this parameter TRUE adjusts the rate of frame changes in an animation to a lower rate if the rendering cannot keep up with the frame changing.','*11','autoFPS','FALSE') newCmd('','11.1.20','','*v+11.2;','Sets the color of all axes. (Same as {#.color~color axes ...}.)','*120','axesColor','"color_name"') newCmd('','11.1.20','','*v+11.2;','Sets the color of the X axis.','*121','axis1Color','"color_name"') newCmd('','11.1.20','','*v+11.2;','Sets the color of the Y axis.','*13','axis2Color','"color_name"') newCmd('','11.1.20','','*v+11.2;','Sets the color of the Z axis.','*14','axis3Color','"color_name"') newCmd('','','','*v+11.4;','Sets a particular model or animation frame to be fixed in place during an {#.animation~} or when displaying models {#.load~loaded} from a multi-model file. set backgroundModel 0 turns this feature off. For a multifile context, the model must be give in "file.model" format. Certain restrictions apply to scripts when a background model is displayed; in that case it may be important to turn the model off and then back on during selected scripting. ','*15','backgroundModel','(integer >= 1) or "file.model"') newCmd('','11.6.RC13','','*v+11.6;','Setting this parameter TRUE allows picking of bonds. If pickCallback is enabled, a message is sent to the callback function providing detailed information about the bond that was picked. See also set atomPicking and set drawPicking.','*17','bondPicking','FALSE') newCmd('','','','*v+11.4;','[NOTE: This setting is deprecated. Starting in Jmol 14.4 you an simply use quotation marks to indicate desired chain case. For example, select :"A" will not select chain a.] Jmol can be set to read the chain designations in PDB, mmCIF, and related files either with or without case sensitivity. With the default set chainCaseSensitive FALSE, the chain designations are interpreted as case-insensitive. With set chainCaseSensitive ON, the chain designation is read in a case-sensitive manner -- chain "A" is different than chain "a". This supports {http://www.rcsb.org/pdb/docs/format/pdbguide2.2/guide2.2_frame.html~PDB format} model files with more than 26 chains. The default startup up mode is OFF -- chain designation "a" in a SELECT command will refer to chain "A" in a file.','*21','chainCaseSensitive','FALSE') newCmd('','14.29.14','','*v+14.4;','Sets {#.calculate~calculate chirality} to apply an advanced CIP Rule 6 that distinguishes "in" from "out" bicyclo fusion stereochemistry.','*22','cipRule6Full','FALSE') newCmd('','14.3.13','','*v+14.4;','Sets the maximum depth of contexts, including {{}}, if{{}}, for{{}}, while{{}}, and function{{}}, as well as the SCRIPT command and a number of expression-related situations. Default is 100; minimum is 10.','*23','contextDepthMax','100') newCmd('','','','*v+11.2;','Sets the default color scheme to be the traditional Rasmol/Chime scheme or the newer, more subtle, Jmol scheme. This command does not actually change the display for an object unless that object is currently being displayed using the default color scheme. See the {http://jmol.sourceforge.net/jscolors/~Jmol Colors page} for default color scheme details.','*31','defaultColorScheme','JMOL or RASMOL') newCmd('','11.3.17','','*v+11.4;','Sets the default scale for the arrow tip for {#.draw} arrows.','*32','defaultDrawArrowScale','(decimal)') newCmd('','','','*v+11.4;','Sets the overall defaults to be Jmol standard or more similar to Rasmol, including default color scheme, axes orientation, zero-based XYZ numbering, no spacefill, and simple wireframe. Applied immediately; should be used prior to file loading.','*330','defaults','JMOL or RASMOL') newCmd('','13.1.16','','*v+13.2;','Sets zoomHeight true and measurementUnits Angstroms','*331','defaults','PyMOL') newCmd('','11.5.12','','*v+11.6;vdw','Sets the default van der Waals radius set for {#.spacefill~} and related commands. For a detailed listing of the values used, see {misc/vdw_comparison.xls~vdw_comparison.xls}. USER refers to a set of data provided by the user using the {#.data~} command.','*34','defaultVDW','JMOL or BABEL or RASMOL or USER') newCmd('','','','*v+11.2;','Setting this parameter TRUEprovides an alternative way to set the default color scheme to RasMol; testable with Jmol math.','*30','colorRasmol','FALSE') newCmd('','11.5.RC3','','*v+11.6;dots','When Jmol displays {#.dots~}, the density of dots is determined by the scale of the display. At a small scale, Jmol may display as few as 12 dots; as zoom increases, this number increases to 42, then 162, and finally, at high zoom, it becomes 642 dots. The dotDensity setting allows control over how many dots are displayed at the various scale level cutoffs. (The actual calculation does not use zoom; rather, it uses a measure of pixels per micron). Setting dotDensity to -3 results in Jmol always displaying 12 dots; setting it to 3 results in Jmol always displaying 642 dots. Settings in between these values decrease or increase the number of dots relative to the default setting (0). ','*351','dotDensity','-3 to 3') newCmd('','12.0.RC24','','*v+12.0;dots;isosurface;mo','Sets the size of {#.dots~} and the dots of {#.isosurface~isosurfaces} and {#.mo~MOs}. ','*352','dotScale','(integer)') newCmd('','','','*v+11.0;dots','Setting this paramter TRUE instructs the {#.calculate~calculate surface} command to disregard atoms that are not selected when calculating the position of the surface (which then determines the parameter {#.atomexpressions~surfaceDistance} for each atom). Also tells Jmol to ignore nonselected atoms when creating a {#.dots~dot surface}. set dotsSelectedOnly TRUE would allow, for example, a continuous set of dots around a ligand even if it is in contact with a protein.','*353','dotsSelectedOnly','FALSE') newCmd('','','','*v+11.0;dots','Setting this parameter OFF instructs Jmol to draw complete spheres of {#.dots~} around each selected atom rather than only the dots that would be derived from that atom in a molecular "dot surface."','*354','dotSurface','ON') newCmd('','','','*v+11.4;','When ON, allows the user to move selected atoms by pressing ALT-SHIFT-LEFT and dragging; when combined with set pickingstyle SELECT DRAG, just LEFT-dragging moves the atoms, and the ALT and SHIFT keys are not required. For a simpler alternative, see {#.set picking~set picking dragSelected} and {#.set picking~set picking dragMolecule}','*401','dragSelected','OFF') newCmd('','13.3.6','','*v+14.0;draw','Sets the font size for {#.draw~} object text.','*411','drawFontSize','14') newCmd('','','','*v+11.4;draw','When ON, and the user hovers over a point associated with {#.draw~} object, the name of the object is displayed next to the point.','*412','drawHover','OFF') newCmd('','','','*v+11.4;draw','When ON, Jmol reports picking of points associated with {#.draw~} objects to the console, the messageCallback function, and the pickCallback function. In addition, setting drawPicking true enables measurement of distances and angles involving drawn objects such as points, lines, arrows, and planes. Measurements of this type only appear transiently; they are not saved. See also set atomPicking and set bondPicking.','*42','drawPicking','OFF') newCmd('','12.3.26','','*v+13.0;','Sets the units of energy for minimization reports in kJ/mol or kcal/mol.','*430','energyUnits','kJ or kcal') newCmd('','11.3.33','','*v+11.4;','Sets (or allows you to inspect, expand, or limit) the list of export drivers available to Jmol. ','*431','exportDrivers','"driver_list"') newCmd('','13.1.19','','*v+13.2;','Sets the export scale for VRML and X3D files created with the {#.write~} command. This setting allows variable scale export to 3D printer CAD software. Defaults to 1.0.','*432','exportScale','(decimal)') newCmd('','12.3.26','','*v+13.0;','Sets the forcefield for minimization to MMFF94 (default) or UFF. If MMFF94 is chosen and any atoms are of a type not recognized by MMFF94, then UFF is used instead.','*441','forcefield','"MMFF" or "UFF"') newCmd('','','','*v+11.0;','Sets the formal charge for the selected atoms.','*442','formalCharge','(integer)') newCmd('','11.8.RC2','','*v+11.8;','Sets the step for calculating straightness and for {#draw~draw helix axis}. The default value is 1, but a value of 2, for example, allows calculating straightness and axes for structures based on pairs of residues. ','*452','helixStep','(integer)') newCmd('','','','*v+11.0;','Sets the web page for the {#.help~} command.','*453','helpPath','"URL"') newCmd('','','','*v+11.2;','Sets the length of time in seconds before a hovering action is acknowledged.','*46','hoverDelay','(decimal)') newCmd('','','','*v+11.2;','Sets the label displayed upon hovering.','*471','hoverLabel','(string)') newCmd('','11.8.RC6','','*v+11.8;','The imageState property, when TRUE, allows Jmol to insert into JPG and PNG files its state. This allows images to serve both as 2D and 3D models.','*472','imageState','ON') newCmd('','','','*v+12.0;','For a multi-touch screen, set this parameter ON if the Jmol applet is running in a browser with the kiosk mode enabled, indicating there are no other applets or applications running. This is a one-time setting -- Setting this parameter OFF is the same as setting it ON, and setting it ON disallows file saves other than logging, prompt dialogs, console, popup menu, and the application script editor. ','*481','isKiosk','OFF') newCmd('','12.2.RC5','','*v+12.2;','Displays a color key relating to the last-generated isosurface. ','*482','isosurfaceKey','OFF') newCmd('','','','*v+11.4;','In the case of an isosurface that is mapped using atom-based property data, the default action is to smooth out the coloring based on an averaging of color weighted by a factor of 1/distance^4 to atoms. Turning this parameter OFF tells Jmol not to do this and instead produce a patchwork mapping that assigns a color based on the property of only the nearest atom. ','*483','isosurfacePropertySmoothing','ON') newCmd('','','','*v+11.4;','When isosurfacePropertySmoothing is set ON, Jmol applies a function to data so as to reduce the harshness of the edges between data assigned to specific atoms. The setting of isosurfacePropertySmoothingPower allows adjustment of that smoothing from 0 (no smoothing) to 10 (strong smoothing). The default value is 7.','*484','isosurfacePropertySmoothingPower','0 to 10 (7)') newCmd('','','','*v+14.4;','Set by Jmol when a state is read that is before 14.2.6 or in the range 14.3.0 - 14.3.5; prevents fractional and cartesian coordinate rounding; used to ensure that states created in JavaScript work identically in Java, and vice versa. Cleared by ZAP or LOAD or loading of any later state script.','*485','jmolInJalview','TRUE') newCmd('','','','*v+14.4;','(Java only) set FALSE for Jmol window to NOT be embedded in JSpecView when JSpecView is opened in Jmol for spectral viewing','*486','legacyJavaFloat','FALSE') newCmd('','','','*v+11.4;','Sets the maximum distance away from a point that that an atom can be found when applying atom data using the {#.load [property]~} command. Default 0.01 Angstroms.Applies the data to all atoms in similar unit cells if the data being read . This allows applying the same data to atoms in all unit cells.','*49','loadAtomDataTolerance','(decimal)') newCmd('','','','*v+11.0;','In situations where there are multiple models, typically only one model is displayed at any given time. In that situation, if a user clicks on a pair of atoms to measure a bond distance, then only one measurement is made. Using set measureAllModels ON, when the user makes measurements in any one frame, ALL similar measurements in all models in hidden frames are generated at the same time. set measureAllModels ON thus allows for very efficient measuring and investigation of differences in a bond distance or bond angle or dihedral angle across multiple models. ','*500','measureAllModels','OFF') newCmd('','12.3.29','','*v+13.0;','Adjusts the width of the lines in isosurface and molecular orbital MESH display','*5011','meshScale','1') newCmd('','','','*v+11.6;','Changes the style of message reporting by script callbacks to one similar to Chime. ','*5012','messageStyleChime','FALSE') newCmd('','','','*v+11.6;','Sets the criterion for stopping a {#.minimize~minimization}. The default value is 0.001; the minimum value is 0.0001.','*5020','minimizationCriterion','(decimal)') newCmd('','14.30.0','','*v+14.30;','Sets the maximum number of atoms that can be minimized using the MMFF-95 force field (default 200).','*5021','minimizationMaxAtoms','(integer)') newCmd('','','','*v+11.6;','Set this flag FALSE to not display {#.minimize~minimizations} as they occur.','*503','minimizationRefresh','TRUE') newCmd('','12.0.RC5','','*v+12.0;','Turns off messages reporting energies for the minimization.','*5041','minimizationSilent','FALSE') newCmd('','','','*v+11.6;','Sets the number of steps after which a {#.minimize~minimization} will stop even if the desired criterion has not been reached. Default value is 100.','*5042','minimizationSteps','(integer)') newCmd('','12.2.RC6','','*v+12.2;','The minimum number of pixels a click can be away from an atom on the screen before it is considered too far away to be a click on that atom.','*5043','minPixelSelRadius','6') newCmd('','14.1.2','','*v+14.2;','Sets the scale of the {#.modulation~} for incommensurately modulated structures. Minimum value is 0.10. ','*5044','modulationScale','(decimal)') newCmd('','','','*v+12.0;','Sets the sensitivity of the mouse when dragging. The default value of 1.0 for set mousedragFactor allows a 180-degrees rotation when the pointer drags across the full window width.','*505','mouseDragFactor','(decimal)') newCmd('','','','*v+12.0;','Sets the sensitivity of the mouse wheel. The default value of 1.0 for set mousedragFactor allows a 180-degrees rotation when the pointer drags across the full window width.','*506','mouseWheelFactor','(decimal)') newCmd('','12.0.RC6','','*v+11.6;','Turns on parallel multiprocessing. If this setting cannot be set true, then it means you do not have a multiprocessor machine. Used in association with the {#.parallelprocess~parallel} and {#.parallel/processs~process} commands.','*508','multiprocessor','FALSE') newCmd('','12.3.3','','*v+13.0;','Determines the URL used for SHOW NMR.','*5091','nmrUrlFormat','"URL"') newCmd('','13.3.9','','*v+14.0;','Sets a global radius for all atoms with SPACEFILL set larger than the max radius value (16.0). Defaults to 20.0. (Any spacefill setting larger than 16 will instead be this setting.)','*510','particleRadius','(decimal)') newCmd('','','','*v+11.6;','Setting this flag TRUE tells Jmol to keep the header portion of the last-loaded PDB file in memory for later retrieval using getProperty("fileHeader"). If FALSE (default), the header is not saved, and a call to this function is slower, since it requires reloading the PDB file and parsing it for its header.','*511','pdbGetHeader','FALSE') newCmd('','','','*v+11.6;','Setting this flag TRUE tells Jmol to assume the groups listed for a given chain in a PDB file are in order already and that Jmol should not check for proper inter-group bonding when assigning polymer sequences that form the basis of secondary structure. This flag allows for custom PDB files where the groups of a chain may not be physically adjacent, yet it is still desired to represent them as single structures.','*512','pdbSequential','FALSE') newCmd('','11.1','','*v+11.2;vdw','The default size of an atom created when a file is loaded as a percent of the atom\'s van der Waal radius (Jmol standard value: 20).','*52','percentVdwAtom','(integer)') newCmd('','11.5.42','','*v+11.6;','Sets the format of the message sent to the console and callback functions when an atom is clicked.','*54','pickLabel','(string)') newCmd('','11.1','','*v+11.2;','The rate of spinning that occurs when a user clicks on the end of a line created using {#.draw~} (default: 20). ','*53','pickingSpinRate','(integer)') newCmd('','11.5.52','','*v+11.2;pg','Sets the tolerance for two atoms being considered identical after application of a rotation. See {#.calculate~calculate pointgroup} for details.','*541','pointGroupDistanceTolerance','(decimal)') newCmd('','11.5.52','','*v+11.2;pg','Sets the tolerance for two axes being considered identical prior to application of a rotation. See {#.calculate~calculate pointgroup} for details.','*543','pointGroupLinearTolerance','(decimal)') newCmd('','11.9.23','','*v+12.0;','This option turns off many memory-consuming features of Jmol that are necessary for preserving the state. It can be used in situations where memory is at a premium and there is no desire to write or save the current Jmol state. ','*550','preserveState','TRUE') newCmd('','','','*v+11.2;','An integer value of 0 or greater. Sets the number of columns to be read for propertyAtomNumberField.','*551','propertyAtomNumberColumnCount','(integer)') newCmd('','','','*v+11.2;','An integer value of 0 or greater. Sets the column (when propertyAtomNumberColumnCount > 0) or free-format field (otherwise) for the atom numbers in a {#.data~} set. These are the atom numbers designated in a PDB file or numbers starting with 1 otherwise. Setting the field to 0 indicates that there is no such field, and values should be read in sequentially.','*552','propertyAtomNumberField','(integer)') newCmd('','','','*v+11.2;','Sets the color scheme associated with properties. SchemeName, in quotes, may be one of "roygb" (default rainbow), "bwr" (blue-white-red), "rwb" (red-white-blue),"low" (red-green), "high" (yellow-blue), "bw" (black-white), "wb" (white-black), and "friendly" (an accessibility option for color-blindness), but may be any user-defined color scheme. In Jmol 11.4, this parameter was largely replaced by color "schemeName".','*56','propertyColorScheme','"colorSchemeName"') newCmd('','','','*v+11.2;','An integer value of 0 or greater. Sets the number of columns to be read for propertyDataField.','*571','propertyDataColumnCount','(integer)') newCmd('','','','*v+11.2;','An integer value of 0 or greater. Sets the column (when propertyAtomNumberColumnCount > 0) or free-format field (otherwise) for the property data in a {#.data~} set. Setting this value to 0 indicates that values are simply to be read sequentially from the data.','*572','propertyDataField','(integer)') newCmd('','11.5.38','','*v+11.6;','Specifies the axes used to define the right-handed {#.quaternion~} frame for proteins and nucleic acids . These frames (xyz axes and origin) define orientations of amino acid residues in proteins and nucleic acid residues in RNA and DNA. The minimum definition of a frame is an origin (only used for {#.draw~draw quaternion}), a point that will define an axis (usually X, but in some cases Y), and a third point that will be used to generate the other two axes. Specifically, if O, A, and B are three points and VOA defines the direction of the X axis, then VC = VOA x VOB defines the direction of the Z axis, and VC x VOA defines the direction of the Y axis. If VOB defines the direction of the Y axis, then VC, as above, defines the direction of the Z axis, and VOB x VC defines the direction of the X axis. Different frames can used for different purposes, ranging from analyzing amino acid side-chain orientations to quantifying the "straightness" of helices and sheets. Most useful are relative and absolute differences of quaternions, which define local helical axes or the axis, angle, and translation required to move from one orientation to another.
[||A| "alternative"| for proteins, same as N (see below)|for nucleic acids, an alternative backbone quaternion with O = P[i], B(Y) = C4\'[i], and A = C4\'[i-1], which is closely associated with eta and theta dihedral angles. ||B| "backbone"| for proteins, O = CA[i], A(X) = CA[i+1], B = CA[i-1]| for nucleic acids, O = P[i], A(X) = P[i+1], B = P[i-1] ||C| "carbon"| for proteins, O = CA, A(X) = C, and B = N, thus defining the orientation of the peptide sidechains|for nucleic acids, same as N, but with the origin shifted to the "center of heavy atoms" for the base (all atoms associated with the base other than H or C4\')||N|"nitrogen"|a frame based on the peptide N atom and useful specifically in the area of solid state NMR spectroscopy. O = N, and if VC = VN-NH, and VD = VN-CA, then VB(Y) = VA x VD, M = axisAngle(VB, -17 degrees), then VA = MVC, and VC = VA x VB defines the direction of the Z axis.|for nucleic acids, O = N9(purines) or N1(pyrimidines), B(Y) = O + VC1\'O, and A = VO-C2 (toward the Watson-Crick edge)||P| "peptide/phosphorus" | for proteins, O = C, A(X) = CA, and Y = N[i+1], thus defining the orientation of the peptide plane| for nucleic acids, O = P[i], A(X) = O3\'[i-1], and B = O5\'[i], thus defining the orientation of the phosphorus tetrahedron||Q|"Quine"| defined as follows: O = (C[i] + N[i+1])/2 and if VX = VCA[i]-C[i] and VB = VN[i+1]-CA[i+1], then VZ = VX x VB, and VY= VZ x VX. This frame was an early definition of the orientation of the peptide plane and suffers from the fact that these two vectors are very nearly colinear, and can produce Z directions that are misdirected; provided for historical reference only (J. R. Quine, Journal of Molecular Structure: THEOCHEM, Volume 460, Issues 1-3, 26 February 1999, pages 53-66).|not used for nucleic acids||RC and RP|"ramachandran"|These frame selections specify that straightness should be calculated from Ramachandran angle approximations rather than actual quaternionFrames "C" and "P", respectively|no nucleic acid equivalent||X|"experimental"|Jmol development testing|Jmol development testing||]','*58','quaternionFrame','A,B,C,N,P,Q,RC,RP,X') newCmd('','','','*v+11.0;','When this flag is set to TRUE, the range for {#color~} temperature is set to be adjustable based on the selected set of atoms being colored; when false (the defualt), the range of colors is set based on the range of values in the entire model. This parameter applies to any property.','*59','rangeSelected','') newCmd('','12.0.RC4','','*v+12.0;','Sets the number of milliseconds to wait before a timer expires signalling that Jmol should not wait any longer for a repaint operation. In some large memory-intensive operations, it is sometimes advisable to set this parameter to a higher number.','*60','repaintWaitMs','1000') newCmd('','11.9.13','','*v+12.0;','Generally when Jmol {#.write~writes} the state, helix/sheet/turn data is saved. In some cases this may not be desired. Setting this flag FALSE prevents the saving of this information in the state.','*61','saveProteinStructureState','TRUE') newCmd('','11.5.52','','*v+11.6;','Generally when selections are made in Jmol all matching atoms in all models are selected, regardless of which model is currently in {#.frame~}. When set FALSE, this flag indicates that the subset of atoms to select should be automatically set to the current model when frame changes occur.','*62','selectAllModels','TRUE') newCmd('','','','*v+11.2;','When turned OFF, selections do not select HETATM atoms in PDB files','*63','selectHetero','ON') newCmd('','','','*v+11.2;','When turned OFF, selections do not select hydrogen atoms','*64','selectHydrogen','ON') newCmd('','','','*v+11.8;','When set TRUE, and {#.set_allowKeyStrokes~} is TRUE, key strokes in the applet or application window to be interpreted as script commands and displayed in the bottom left corner of the window.','*650','showKeyStrokes','TRUE') newCmd('','','','*v+14.4;','Set this value TRUE if VECTORS command is to show modulation displacements instead of vibrations','*651','showModulationVectors','FALSE') newCmd('','13.1.6','','*v+13.2;','When set TRUE, reports timings for isosurface creation in ms in the Jmol console, and in the Java console reports detailed timings for rendering of shapes.','*66','showTiming','FALSE') newCmd('','12.1.10','','*v+12.2;','Sets a slab range that is independent of zoom. ','*67','slabRange','(decimal)') newCmd('','11.3.29','','*v+11.2;','Turning the smartAromatic parameter OFF reverts to a Jmol 10 style of drawing aromatic bonds as a paired solid and dashed line when loading subsequent files. (This command has no immediate effect. Use {#.calculate~reset aromatic;calculate aromatic} to see the effect of smartAromatic)','*68','smartAromatic','ON') newCmd('','','','*v+11.2;','determines the number of frames per second between displays of the molecule -- a small number here results in a jerky stepwise rotation. ','*82','spinFps','._spin_fps') newCmd('','','','*v+11.2;','The set spinX, set spinY, and set spinZ commands allow for the setting of the spin axes -- x, y, and z -- independently as well as the rate of spin. The actual spinning axis is a complex combination of the three settings. No actual spinning occurs until the {#.spin~spin ON} command is issued or the user turns spinning on using the mouse menu. Jmol has a much richer variety of spinning possibilities to this Chime/Rasmol-standard capability, allowing simple spinning around arbitrary molecular- and window-based axes. See the {#.rotate~} command.','*84','spinX','._spin_rate') newCmd('','','','*v+11.2;','see set spinX','*86','spinY','._spin_rate') newCmd('','','','*v+11.2;','see set spinX','*87','spinZ','._spin_rate') newCmd('','14.1.16','','*v+14.2;','Sets the star line width in Angstroms.','*881','starWidth','(decimal)') newCmd('','','','*v+11.2;','When set OFF, this parameter prevents Jmol from recording status messages and reporting them to a requesting web page. When OFF, the JavaScript method Jmol.scriptWait() will function correctly, but it will not return status information from the applet.','*891','statusReporting','ON') newCmd('','','','*v+11.0;','Same as {#.stereo~stereo (degrees)}, sets the angle for stereo images; can be checked using Jmol math.','*893','stereoDegrees','(decimal)') newCmd('','11.3.57','','*v+11.4;','For {#.calculate~calculate STRUTS} and the {#.struts~} command, sets the default radius for new struts. ','*8941','strutDefaultRadius','0.3') newCmd('','11.3.57','','*v+11.4;','For {#.calculate~calculate STRUTS}, sets the maximum length for a new strut. ','*8942','strutLengthMaximum','7') newCmd('','','','*v+11.2;','For {#.calculate~calculate STRUTS}, turn this flag ON to allow more than one strut on a given atom. ','*8943','strutsMultiple','OFF') newCmd('','','','*v+11.0;','For {#.calculate~calculate STRUTS}, sets the minimum spacing (in terms of residues) for new struts. ','*8944','strutSpacing','6') newCmd('','11.3.57','','*v+11.4;','When {#.sync~} is ON, delivers mouse actions to the target applet.','*894','syncMouse','OFF') newCmd('','11.3.57','','*v+11.4;','When {#.sync~} is ON, delivers script events to the target applet.','*895','syncScript','OFF') newCmd('','','','*v+12.0;','Setting this flag FALSE disables the undo/redo buttons on the Jmol application console, saving memory.','*901','undo','ON') newCmd('','11.7.40','','*v+11.8;','Generally the {#.minimize~} command runs in a separate thread. Setting this parameter FALSE instructs Jmol to use the same thread as is being used for commands. This is important in situations where one wants to wait for completion of the minimization before continuing.','*905','useMinimizationThread','ON') newCmd('','','','*v+11.0;','Currently only applicable in the display of unit cell parameters, the useNumberLocalization setting determines whether or not local number formatting will be used (comma as decimal point, for example).','*91','useNumberLocalization','ON') newCmd('','','','*v+11.0;','Sets the scale for vibration vectors.','*920','vectorScale','(decimal)') newCmd('','','','*v+14.6;','For spinning magnetic vectors, draw a Japanese fan-like trail for the specified number of animation frames.','*921','vectorTrail','(decimal)') newCmd('','14.1.16','','*v+14.2;','Sets vectors to be centered on an atom (as for showing magnetic spin vectors).','*930','vectorsCentered','') newCmd('','','','*v+12.2;','Sets the default VDW radius to be based on Jmol values (OpenBabel or Rasmol, depending upon file type) or the set used by the Duke University {http://molprobity.biochem.duke.edu/~molProbity} site as per Word, et al, J. Mol. Biol. (1999) 285, 1711-1733: [|| 1.0 | {{_H}}|| 1.17 | {{_H and connected(_C) and not connected(within(smiles,"[r6]"))}}|| 1.75 | {{_C}}|| 1.65 | {{_C and connected(3) and connected(_O)}}|| 1.55 | {{_N}}|| 1.4 | {{_O}}|| 1.8 | {{_P}}|| 1.8 | {{_S}} ||]','*931','vdw','JMOL or PROBE') newCmd('','12.3.2','','*v+13.0;','Displays vibration vectors as double-ended arrows.','*932','vectorSymmetry','FALSE') newCmd('','','','*v+11.0;','Sets the default period of vibrations (in seconds). Setting this value to 0 disables vibration modeling.','*933','vibrationPeriod','(decimal)') newCmd('','','','*v+11.0;','Sets the amplitude of vibrations.','*95','vibrationScale','(decimal)') newCmd('','11.9.24','','*v+12.0;','Setting this parameter OFF allows {#.moveTo~} operations to continue independently while script commands are processing. An ongoing moveTo operation can then be stopped at any time using moveTo STOP Starting in Jmol 14.2.7, this action is extended to the {#.rotate~rotate x.x y.y} finite spin option.','*96','waitForMoveto','ON') newCmd('','11.3.55','','*v+11.4;','Turning this parameter ON sets Jmol to not display spacefill and only display bonds as simple lines during user-based model manipulation. ','*97','wireframeRotation','OFF') newCmd('.set modelKitMode','12.0.RC15','','*v+12.0 -- new modelKitMode;','Jmol includes a mode that allows building and modifying models, with minimization. Features include the ability to easily add, drag, and delete atoms, drag and minimize molecules and molecular fragments such as CH3 groups, rotate bonds, minimize parts of models -- all mouse-driven. A special menu is employed that sits at the top left corner of the Jmol window. (Clicking the Jmol frank still brings up the standard menu.) After set modelKitMode, a {#.zap~} command creates a CH4 model, and the picking mode goes to set picking assignAtom_C. ','1|2','','') newCmd('.set picking','','','*v+12.0 -- adds set picking measure SEQUENCE;','The set picking command determines the response to clicking of atoms by the user. For those options for which they apply, the keywords MEASURE and SELECT are optional.','2|2','','') newCmd('','','','','Setting this paramter OFF turns off the sending of picking information (atom identifier, atom number, and atomic position) to both to the status line and to the {#.set_pickCallback~PickCallback} function, if defined.','*1','ON','') newCmd('','','','','When the user clicks an atom, that atom is set to be the center of rotation. If windowCentered is true (default), then this atom is smoothly moved to the window center; if not windowCentered, then the atom stays in position, but now all rotation is around it and if perspectiveDepth is set (set windowCentered off;set perspectiveDepth on), the perspective around this atom shifts to indicate that it is now the focus of the perspective. If the same atom is clicked twice, it is zoomed in on by a factor of two. If no atom is clicked, then the model is zoomed out by a factor of two. ','*4','CENTER','') newCmd('','11.9.21','','','Performs like measuring distances, except bonds are created. ','*50','CONNECT','') newCmd('','11.9.28','','','Clicking on an atom deletes it. ','*51','DELETEATOM','') newCmd('','11.9.21','','','Deletes the bond between pairs of clicked atoms (and with set BONDPICKING TRUE, deletes bonds as they are clicked.. ','*52','DELETEBOND','') newCmd('','12.0.RC15','','','Clicking and dragging an atom moves its position.','*54','DRAGATOM','') newCmd('','13.1.17','','*v+13.2;','Clicking and dragging an atom of a ligand moves its position. LEFT-drag translates; ALT-LEFT-drag rotates. Similar to set picking dragMolecule except PDB non-ligand groups are not moved. ','*55','DRAGLIGAND','') newCmd('','12.0.RC15','','','Clicking and dragging an atom moves its position and then does a minimization of its associated molecule.','*56','DRAGMINIMIZE','') newCmd('','12.0.RC15','','','Clicking and dragging an atom moves and minimizes its associated molecule. This setting can be used to simulate docking. In the case of clicking on a protein side chain, that side chain will be minimized. It is important to use set pdbAddHydrogens prior to loading the protein in order to avoid a stereochemical inversion with certain sidechains such as ILE or THR, or be very careful about that. ','*57','DRAGMINIMIZEMOLECULE','') newCmd('','','','*v+11.4;','Clicking and dragging on any atom of the structure drags its molecule only; using SHIFT with the drag rotates the molecule about its center.','*581','DRAGMOLECULE','') newCmd('','12.1.46','','*v+12.2;','Clicking and dragging on any atom of the structure drags the selected atoms only; using SHIFT with the drag rotates the selected atoms about their center. See also {#.set_allowmoveatoms~set allowMoveAtoms} and {#.set_allowrotateselected~set allowRotateSelected}, both of which modify the action of set picking DRAGSELECTED.','*582','DRAGSELECTED','') newCmd('','','','','The vertices of all {#.draw~} objects are highlighted. Holding the SHIFT key down while dragging moves the object to a new location ("shifts" the object); holding ALT down while dragging moves a single vertex to a new location ("alters" the shape of the object). The draw command required to reproduce the object can be seen immediately using the Java console. This new command can also be seen via message callback, or in the Jmol {#.console~} using {#.show~show DRAW}.','*63','DRAW','') newCmd('','','','','Same as set picking ON.','*65','IDENTIFY','') newCmd('','12.0.RC15','','*v+12.0;','Selecting an atom that is part of a ring after set picking invertStereo will reverse the two non-ring atoms -- actually rotating them 180 degrees, not doing a planar inversion, thus preserving whatever chirality might be attached to them. (See also {#.invertSelected~invertSelected STEREO}.','*67','INVERTSTEREO','') newCmd('','','','','When the user clicks an atom, the label of that atom is toggled on or off. Pressing SHIFT while dragging a label changes its position relative to the atom.','*69','LABEL','') newCmd('','','','','Same as set picking MEASURE DISTANCE but also displays a distance measurement on the molecule.','*70','MEASURE','') newCmd('','','pickd','','Turns picking on and returns atom identities and distance between two atoms. Three messages are sent to the {#.set_messageCallback~MessageCallback} function, if defined: Atom #1 (after the first click) and then Atom #2 and Distance (after the second click).','*71','MEASURE','DISTANCE') newCmd('','','picka','','Turns picking on and returns atom identities and angle involving three atoms. Four messages are sent to the {#.set (callback)~MessageCallback} function, if defined: Atom #1 (after the first click), Atom #2 (after the second click), and then Atom #3 and Angle (after the third click).','*72','','ANGLE') newCmd('','','pickt','','Turns picking on and returns atom identities and torsion angle (dihedral angle) involving four atoms. Five messages are sent to the {#.set_messageCallback~MessageCallback} function, if defined: Atom #1 (after the first click), Atom #2 (after the second click), Atom #3 (after the third click), and then Atom#4 and Torsion (after the fourth click).','*74','','TORSION') newCmd('','','','*v+12.0;','Turns picking on and returns the 1-letter-code sequence of atoms in bioSMILES format, showing chain and residue range and biomolecular type (p for protein, d for dna, r for rna). For example: //* chain A protein 36 *// ~p~PG //* 37 *//.','*75','','SEQUENCE') newCmd('','','','','User mouse clicking sets the navigation center to the selected position. See {#.navigation}.','*83','NAVIGATION','') newCmd('','','','','When the user clicks an atom, the selection of that atom is toggled on or off.','*841','SELECT','ATOM') newCmd('','','','','When the user clicks an atom, the selection of the chain associated with that atom is toggled on or off. ','*842','','CHAIN') newCmd('','','','','When the user clicks an atom, the selection of the group associated with that atom is toggled on or off.','*843','','GROUP') newCmd('','','','','When the user clicks an atom, the selection is toggled on or off for all VISIBLE atoms with the same element as the selected atom.','*844','','ELEMENT') newCmd('','11.9.24','','*v+12.0;','When the user clicks an atom, the selection is toggled on or off for all atoms within the same polymer unit (PDB only).','*846','','POLYMER') newCmd('','11.9.24','','*v+12.0;','When the user clicks an atom, the selection is toggled on or off for all atoms within the same structural unit (helix, sheet, turn -- PDB only).','*89','','STRUCTURE') newCmd('','','','*v+11.0;','When the user clicks an atom, the selection is toggled on or off for all atoms within the same covalenty bonded set as the selected atom.','*845','','MOLECULE') newCmd('','','','*v+11.0;','When the user clicks an atom, the selection is toggled on or off for all VISIBLE atoms with the same crystallographic site as the selected atom.','*85','','SITE') newCmd('','','','*v+11.0;','Turns picking on and sets it so that when the user clicks two atoms in succession, the model starts to spin around the axis defined by those two atoms. A third click on any atom stops rotation. Optionally, a speed of rotation can be included.','*90','SPIN','._spin_fps') newCmd('','','','*v+11.0;','When the user clicks two atoms, a {#.struts~strut} is created between them.','*93','STRUTS','') newCmd('','','','*v+12.0;','When the user clicks two atoms of a crystallographic file, a representation of their symmetry relationship is drawn, and the operator(s) relating these positions is written to the console. ','*94','SYMMETRY','') newCmd('.set pickingStyle','','','*v+11.0 -- NEW;','With set pickingStyle you can change the behavior of Jmol in response to user mouse actions relating to selecting atoms or measuring distances, angles, and torsions.
[||SELECT|Sets the behavior of Jmol to different clicking styles. For the standard Jmol default selection behavior, use set pickingStyle SELECT toggle The actions of these options depend upon the setting of {#.set picking~set picking SELECT}. If picking has not been set to SELECT, then this command has no immediate effect. In the explanations given below, it is presumed that we have set picking SELECT GROUP. The SELECT keyword is recommended but not required.||MEASURE|set pickingStyle MEASURE ON in conjunction with set picking MEASURE displays the measurements on the structure as they are generated by user clicking. If picking has not been set to MEASURE, then this command has no immediate effect. set pickingStyle MEASURE OFF returns to the default Jmol behavior.||]','0|1','','') newCmd('','','','*v+11.0;','left-click toggles that group selected/not selected (Chime style; Jmol default).','*1','SELECT','toggle') newCmd('','','','*v+11.0;','left-click selects just that group
shift-left-click toggles the group selected/not selected (Rasmol style).','*2','','selectOrToggle') newCmd('','','','*v+11.0;','User mouse action is according to the following scheme, which is the style used by {http://pfaat.sourceforge.net~PFAAT}.
[||left-click |selects just that group, like rasmol||shift-left-click|toggles the group selected/not selected||alt-left-click |appends the group to the current selection||alt-shift-left-click|removes the group from the current selection||left-click off model |executes (select none) ||]','*3','','extendedSelect') newCmd('','11.9.24','','*v+12.0;','makes the LEFT button a click-and-drag button when associated also with set PICKING select (molecule, group, chain, etc.) and set dragSelected.','*4','','DRAG') newCmd('','','','*v+11.0;','Returns to the Jmol default toggle picking style.','*5','','NONE') newCmd('','','','*v+11.0;','Clicking atoms measures and displays distance, angle, or torsions based on the setting of set picking MEASURE. By default the user sees this information displayed. Setting set pickingStyle measure OFF when set picking MEASURE is set to DISTANCE, ANGLE, or TORSION tells Jmol to stop indicating measurements on the model when the user clicks, even though the distance, angle, or torsion information is still sent to the message queue. ','*6','MEASURE','ON') newCmd('.show','','','*v+13.0 -- adds show chemical, DSSP, NMR;','show sends information about the model to the {#.set (callback)~MessageCallback} function and to the Java or Jmol {#.console~}. If using the application, using Jmol -ionx filename.spt modelfile.xyz > output.txt, you can put a show command in a script file (filename.spt), and have the output directed to output.txt. The command line options used in this example include -i (silent startup), -o (use standard output), -n (no display), -x (exit after running the specified script file). All of the parameters that can be {#.set~} can be shown. They are not listed here. Adding "/xxxx" to the command, such as SHOW file/cell will filter the output only to lines containing the text after the slash character.','0|1','','') newCmd('.show','','','*v+11.6 -- adds show moveto, show rotation, show translation, show atom, show chain, show group, show selected;*v-13.0;','show sends information about the model to the {#.set (callback)~MessageCallback} function and to the Java or Jmol {#.console~}. If using the application, using Jmol -ionx filename.spt modelfile.xyz > output.txt, you can put a show command in a script file (filename.spt), and have the output directed to output.txt. The command line options used in this example include -i (silent startup), -o (use standard output), -n (no display), -x (exit after running the specified script file). All of the parameters that can be {#.set~} can be shown. They are not listed here.','0|1','','') newCmd('','','','*v+11.0;','Displays the {misc/JVXL-format.pdf~JVXL} file equivalent. (Not available for all object types.)','*4','ISOSURFACE','') newCmd('','11.5.45','','*v+11.6;','Lists the atom numbers that are currently selected in the order of atom index; duplicates are not listed twice.','*111','ATOM','') newCmd('','13.3.6','','*v+14.0;','Operates on the currently selected atoms, reporting a quarternion that would rotate them to their {#.rotate_best~best} orientation for viewing. ','*121','BEST','ROTATION') newCmd('','13.3.6','','*v+14.0;','Operates on the currently selected atoms, reporting the volume and dimensions of the best box that would contain the selected atoms. ','*122','BEST','BOX') newCmd('','','','centering;boundbox','Delivers the coordinates of the center coordinate and a directional vector defining a box just perfectly enclosing the model. The vector is determined by taking the min and max values for the atom along each cartesian axis. The center is the geometric center of the model, not the default center of rotation (which is the mean atom position). The eight corners of the boundbox are found by adding the center point to the vector, with all possible combinations of +/- component cectors. The length of a side of the boundbox is determined by doubling the appropriate component of the vector. So, for example, the length of the boundbox along the x-axis is (2*vectorX). Units are in Angstroms. Output is in the form
boundbox: (centerX, centerY, centerZ) (vectorX, vectorY, vectorZ)','*13','BOUNDBOX','') newCmd('','13.1.2','','*v+13.2;cache','Lists the contents of the {#.cache~}.','*19','CACHE','') newCmd('','','','centering','Delivers the coordinates of the center of the model. Units are in Angstroms. Output is in the form
center: (centerX, centerY, centerZ)','*20','CENTER','') newCmd('','11.5.45','','*v+11.6;','Lists the chains (A, B, C, etc.) of the atoms that are currently selected in the order of atom index; duplicates are not listed twice.','*21','CHAIN','') newCmd('','12.1.43','','*v+12.2;','Displays a property of the selected atoms depending upon the selected file type option obtained from the {https://cactus.nci.nih.gov~NIH Catus server}. These include: alc ,cdxml, cerius, charmm, cif, cml, ctx, gjf, gromacs, hyperchem, jme, maestro, mol, mol2, sybyl2, mrv, pdb, sdf, sdf3000, sln, and xyz. In addition to these, the SHOW command also allows for show SMILES (created by Jmol) and show DRAWING (same as show CHEMICAL IMAGE). [||cas|CAS Registry Number||chemspider_id|ChemSpider ID||ficts|FICTS Identifier||ficus|FICuS Identifier||formula|Chemical Formula||effective_rotor_count|Number of Effectively Rotatable Bonds||hashisy|Cactvs HASHISY||h_bond_donor_count|Number of Hydrogen Bond Donors||h_bond_acceptor_count|Number of Hydrogen Bond Acceptors||h_bond_center_count|Number of Hydrogen Bond Acceptors and Donors||image|GIF Image||inchi| {http://www.iupac.org/inchi~InChI} string||inchikey|InChI key||iupac_name|IUPAC Name||mw|Molecular Weight||name|the first name of the compound found in the NIH database||names|list of known names of the compound, if found in the NIH database||ring_count|Number of Rings||ringsys_count|Number of Ring Systems||rotor_count|Number of Freely Rotatable Bonds||rule_of_5_violation_count|Number of Rule of 5 Violations||smiles|SMILES||stdinchi|standard {http://www.iupac.org/inchi~InChI} string||stdinchikey|standard InChI key||uuuuu|uuuuu Identifier||]','*221','CHEMICAL','[option]') newCmd('','11.3.13','','*v+11.4;color','Displays the list of colors comprising a color scheme, where "name" is, for example, "roygb", "rwb", or "user", "byElement_Jmol", "byElement_Rasmol", "byResidue_Shapely" (cooresponding to color shapely), and "byResidue_Amino" (corresponding to color amino). ','*222','COLORSCHEME','"name"') newCmd('','','','*v+11.0;','Displays the most recently read {#.data~} of the given type. See also {#.getProperty~getProperty data}.','*25','DATA','"type"') newCmd('','14.2.6','','*v+14.4;','Shows {#.annotationssequencedomainannotations~domain annotations} that have been loaded from RCSB or PDBe with the /dom option. For example,
load =1crn/dom;show domains SCOP
1crn SCOP 57430 identifier=Crambin-like
1crn SCOP 57430 fold description=Crambin-like
1crn SCOP 57430 fold sunid=57428
1crn SCOP 57430 description=Crambin-like
1crn SCOP 57430 class description=Small proteins
1crn SCOP 57430 class sunid=56992
1crn SCOP 57430 superfamily description=Crambin-like
1crn SCOP 57430 superfamily sunid=57429
','*302','DOMAINS','') newCmd('','12.1.43','','*v+12.2;','Displays a drawing of the Jmol structure in a new browser window.','*305','DRAWING','') newCmd('','','','','Displays the command required to generate the current {#.draw~} object. Particularly useful after using {#.set picking~set picking DRAW}.','*303','DRAW','') newCmd('','12.0.18','','*v+12.2;','Displays a DSSP {misc/1gua-dssp-jmol.txt~secondary structure analysis report}, which includes regions of three kinds of helix (3/10, alpha, and pi), turns, sheets, and bridge groups (DSSP codes G, H, I, T, E, and B, respectively). Bends (DSSP "S") are not included. This report can be sent to a file using, for example: var x = script("show DSSP");write var x "dsspReport.txt".','*31','DSSP','') newCmd('','','','','Delivers the entire contents of the file for the current model. ','*32','FILE','') newCmd('','','','','Delivers the entire contents of the specified file on the server from which the applet was loaded. The filename must be relative to the current page (not necessarily the directory containing the applet) and must be enclosed in quotation marks. If the file is in ZIP format -- JAR, ZIP, JMOL -- or contains ZIP data (PNGJ), only a directory of the contained files is reported. ','*33','FILE','filepath') newCmd('','11.3.23','','*v+11.4;','Lists all user-defined functions.','*53','FUNCTIONS','') newCmd('','11.5.45','','*v+11.6;','Lists the groups (ALA, VAL, etc.) of the atoms that are currently selected in the order of atom index; duplicates are not listed twice.','*54','GROUP','') newCmd('','','','*v+11.0;','Shows n lines of command history. If no number is given, show history by itself shows the full set of recorded command lines (100 maximum by default, but this maximum can be changed using the {#.set(files and scripts)~set history} command. History recording can be turned on and off using the {#.history~} command.','*63','HISTORY','n') newCmd('','12.1.51','','*v+12.2;','Displays lighting details, including ambientPercent, diffusePercent, specular, specularPercent, specularPower, specularExponent, and zShade','*68','LIGHTING','') newCmd('','','','*v+11.0;','Displays a table of information relating to {#.measure~measurements} that have been made.','*70','MEASUREMENTS','') newCmd('','','','*v+11.4;','Displays the current Jmol menu in a format that can be loaded using {#.load MENU~}.','*710','MENU','') newCmd('','','','*v+11.4;minimize','Displays a detailed report regarding minimization parameters used for the most recent {#.minimize~minimization}.','*711','MINIMIZATION','') newCmd('','','','*v+11.0;','Displays the {misc/JVXL-format.pdf~JVXL} file equivalent for the current {#.mo~molecular orbital}. If no MO is currently shown, displays the JVXL equivalent for the entire set of molecular orbitals. (This can take some time to construct.)','*72','MO','') newCmd('','','','','Delivers properties associated with the currently loaded model. Output includes information about all of the models in the set. This command is still in development. The exact form and content of the output is subject to change (and suggestion).','*73','MODEL','') newCmd('','11.5.45','','*v+11.6;','Same as show orientation moveto','*75','MOVETO','') newCmd('','12.3.3','','*v+13.0;','Links to {http://www.nmrdb.org/predictor~} (or whatever URL is indicated in set nmrUrlFormat using the SMILES string of the selected atoms. ','*76','NMR','') newCmd('','','','','Delivers the orientation of the model. Output consists of both a "moveTo" command and an alternative sequence of "reset; rotate z; rotate y; rotate z" commands that would result in the current orientation. Thus, this command allows reading and restoring specific user-specified orientations. Optional types include: [||moveto|the moveto command leading to this orientation, with no comments ||rotation|the current rotation, as a {#.quaternion~}||translation|the current translation in percent X and percent Y from center of the window||]','*77','ORIENTATION','[optional type]') newCmd('','','','','Delivers the PDB file header.','*831','PDBHEADER','') newCmd('','11.5.52','','pg','Displays a table summarizing the point group of a symmetrical or nearly symmetrical model. See {#.calculate~calculate pointgroup} for details of this calculation.','*833','POINTGROUP','') newCmd('','14.3.15','','*v+14.4;pg,poly','Displays a table summarizing the point group of a selected polyhedron. One atom (one polyhedron) should be selected prior to this command.','*834','POINTGROUP','POLYHEDRON') newCmd('','14.3.15','','*v+14.4;','Displays a list of up to three atom properties, optionally along with atom name and index. For example, show properties x y z tabulates just x, y, and z coordinate values. Adding a format allows rounding and formatting as for labels. Two special formats, %s and %i, add atom name and index, respectively. For example, show properties x y z FORMAT "%s %i %10.6f %10.6f %10.6f". The corresponding WRITE command puts these into a file.','*835','PROPERTIES','(list)','FORMAT','"%s %i ..."') newCmd('','11.5.39','','*v+11.6;','Reports a listing of currently selected residues:
[GLY]1:A[ARG]2:A','*841','RESIDUES','') newCmd('','11.5.45','','*v+11.6;','Same as show orientation rotation','*843','ROTATION','') newCmd('','11.5.39','','*v+11.6;','Reports the sequence of the currently selected residues in the following format:
[ARG]3:A
[ILE]4:A
[GLN]5:A
[GLY]6:A
[GLN]7:A
[ARG]8:A
[ARG]9:A
[GLY]10:A
[ARG]11:A
[GLY]12:A
Model 1.','*852','SEQUENCE','') newCmd('','11.5.45','','*v+11.6;','Lists the default label for all selected atoms, in the order of atom index.','*851','SELECTED','') newCmd('','','','*v+11.2;','Delivers a list of all "set" commands that have been issued, with their values.','*861','SET','') newCmd('','','','*v+12.0;','Delivers a SMILES string for the selected atoms. Optional directives include /strict, /open, and /nci.','*863','SMILES','/directive') newCmd('','','','*v+11.0;','Displays information relating to crystallographic space groups. If a file with crystallographic symmetry is loaded, show spacegroup by itself displays information for that spacegroup. Quotes are required around the space group name. If the name itself includes double quotes, use two single quotes instead. For example: P 32 2\'\', not P 32 2". ','*87','SPACEGROUP','"name"') newCmd('','','','*v+11.0;','Delivers a Jmol script that, when run, will regenerate the state of the Jmol applet or application. The following objects are not yet supported: dipole, isosurface, lcaoCartoon, mo. If a name is given, then the state {#.save~saved} with that name is used; if no name is given, the state described is the current state.','*880','STATE','[optional name]') newCmd('','12.1.14','','*v+12.2;','Shows only the lines from the current state that contain the specified phrase "xxx"','*881','STATE','/xxx') newCmd('','12.1.48','','*v+12.2;','Delivers the Jmol state embedded in an image file of the format PNG or JPG created by Jmol (and thus containing the string "**** Jmol Embedded Script ****"). ','*882','STATE','FILE','"fileName"','') newCmd('','14.1.16','','*v+12.0;draw','Describes the specified symmetry operation. For example, show symop 3 might give "-y,x-y,z-1/3 3-fold screw axis|translation: 0 0 -1/3". If no integer is given, this command delivers the full list of symmetry operations for the current frame. Starting with Jmol 14.2, an optional additional flag "fmatrix" reports the symmetry operation as a matrix rather than in Jones-Faithful notation.','*891','SYMOP','(integer)') newCmd('','14.1.16','','*v+12.0;draw','Describes the symmetry operation relating the two specified points. (If an atom set is given, only the average position of the coordinates is used.) For example, show symop {{molecule=1}} {{molecule=2}} might give "3 -y,x-y,z-1/3 3-fold screw axis|translation: 0 0 -1/3". If more than one symmetry operation relates the positions, then all are described. Starting with Jmol 14.2, an optional additional flag "fmatrix" reports the symmetry operation as a matrix rather than in Jones-Faithful notation.','*892','SYMOP','._atom_expression_or_coord','._atom_expression_or_coord','') newCmd('','11.9.17','','*v+12.0;draw','Shows the symmetry relation associated with the specified symmetry operator between any two atoms or groups or any two positions in space. For example, load =ams/quartz 1 packed;show symop 2 @1 @3.','*894','SYMOP','(integer)','{{atom expression}}','{{atom expression}}') newCmd('','11.9.17','','*v+12.0;draw','Shows the nth symmetry operator for special positions, where multiple symmetry operations relate two positions in space. For example, load =ams/quartz 1 packed;show symop @1 @3 1.','*895','SYMOP','{{atom expression}}','{{atom expression}}','(integer)') newCmd('','11.9.3','','*v+12.0;draw','Shows the symmetry operation associated with the given 4x4 matrix, which most likely comes from a variable, for example here the symmetry operation that is the product of two other symmetry operations: show SYMOP @{{symop(11)*symop(14)}}.','*896','SYMOP','[matrix]') newCmd('','','','*v+11.0;','Displays information relating to the crystallographic symmetry of the model, including space group name and symmetry operations. (Applicable file formats only.)','*897','SYMMETRY','') newCmd('','11.9.10','','*v+12.0;','Lists all pending {#set(timeout)~timeouts}.','*901','TIMEOUT','') newCmd('','','','*v+11.8;','Reports a trace of function and script calls leading to the current command.','*902','TRACE','') newCmd('','','','','Delivers the current 3x3 transformation matrix (rotation only). ','*903','TRANSFORM','') newCmd('','11.5.45','','*v+11.6;','Same as show orientation translation','*905','TRANSLATION','') newCmd('','','','*v+11.0;','Displays information relating to the crystallographic unit cell of the model. (Applicable file formats only.)','*91','UNITCELL','') newCmd('','','','*v+11.0;','Opens the data file in a new browser window. (applet only)','*92','URL','') newCmd('','','','*v+11.0;','Opens the specified URL in a new browser window. (applet only)','*93','URL','URL') newCmd('','14.2.6','','*v+14.4;','Shows {#.annotationsstructurevalidationannotations~validation annotations} that are available as properties, having been loaded from RCSB or PDBe with the /val option. For example,
Chain A:
[GLY]1 [ARG]2 [ARG]3 [ILE]4 [GLN]5
[GLY]6 [GLN]7 [ARG]8 [ARG]9 [GLY]10
[ARG]11 [GLY]12 [THR]13 [SER]14 [THR]15
[PHE]16 [ARG]17 [ALA]18 [PRO]19 [SER]20
[HIS]21 [ARG]22 [TYR]23 [LYS]24 [ALA]25
[ASP]26 [LEU]27 [GLU]28 [HIS]29 [ARG]30
[LYS]31 [VAL]32 [GLU]33 [ASP]34 [GLY]35
[ASP]36 [VAL]37
load *1crn/val; show validation
Validations loaded:
property_types_of_outliers (residues: 3)
property_bond_lengths (atoms: 2, max: 5.38)
property_bond_angles (atoms: 6, max: 7.83)
','*948','VALIDATION','') newCmd('','','','*v+11.8;','Reports a listing of all variables and their values.','*949','VARIABLES','') newCmd('','','','','Delivers the current zoom setting. Output is in the form of the zoom command: "zoom n" where "n" is an integer percent of "normal" zoom.','*95','ZOOM','') newCmd('','','','*v+11.0;','For {#.draw~} objects, displays the command that can be used to generate that object. For {#.isosurface~} objects, displays the {misc/JVXL-format.pdf~JVXL} file equivalent. The $ is required. (Not available for all object types.)','*99','$objectID','') newCmd('.show (NMR)','13.3.6','','*v+14.0;nmr','Displays a predicted 1H NMR spectrum using JSpecView (Jmol application only). Atoms on the structure are correlated with signals in the spectrum. The spectrum is constructed using a service provided by {http://nmrdb.org~}.
 ','0|1','','')
newCmd('.slab','11.1.5','','*v+11.2 adds internal slabbing;slabdepth;disp;misc',' Slab and Depth together control the percentage of the molecule to be displayed based on clipping planes. All of the commands described here for {#.slab~} are also available for {#.depth~}. slab on turns slab/depth on. slab 50 shows the back 50% of the molecule. slab 25 show the back 25% of the molecule. depth does the opposite of slab, hiding atoms far from the user. The default settings to see all of the model, then, are slab 100; depth 0. Atoms appear solid; bonds appear hollow. Slabbing can also be applied "internally" -- that is, based on molecular coordinates, not screen coordinates. Internal slabbing involves defining a plane ax + by + cz + d = 0 as {{a b c d}}, using Miller indices {{h k l}}, or using standard notation such as "x=3" or "xy". CHIME NOTE: The slab/depth effect is equivalent to the RasMol command \'set slabmode solid\', however \'set slabmode [option]\' is not supported. ','0|1','','')
newCmd('','','','','Turns slab/depth on or off. Either slab ON or depth ON can be used; in either case both slab and depth are turned on.','*1','._on_off{"ON"}','')
newCmd('','','','','Sets the position of the slab/depth plane from 0 (front) to 100 (rear) of the model','*2','._percent_slab','')
newCmd('','11.1.5','','*v+11.2;','Sets the position of the slab/depth plane based on the specified Miller index plane. For slabbing, with positive hkl values, atoms far from {{0 0 0}} are removed. The value NONE removes the slab/depth plane','*32','HKL','{{h k l}} or NONE')
newCmd('','11.1.5','','*v+11.2;','Sets the position of the slab/depth plane based on the specified Miller index plane; reference point is opposite slab hkl or depth hkl.','*33','-HKL','{{h k l}}')
newCmd('','11.1.5','','*v+11.2;','Sets the slab/depth plane based on a plane using the general syntax for {#.plane expressions~}. For example: slab PLANE {{atomno=3}} {{atomno=2}} {{atomno=1}}; slab on. Note that the order of points defines the direction from the plane of atoms to be removed. If using your right hand to define the path from first point to second to third, your right thumb points to the atoms removed by slabbing. (Opposite this for depth.)','*41','PLANE','plane_expression or NONE')
newCmd('','11.1.5','','*v+11.2;','Reverses the sense of the plane defined as above.','*42','-PLANE','plane_expression')
newCmd('','11.1.5','','*v+11.2;','Resets slab and depth to the standard slab 100, depth 0, clears any internal planes, and turns slab on.','*7','RESET','')
newCmd('','11.1.5','','*v+11.2;','Sets the current slab (using slab on) to be internal, so that rotation of the model preserves the current slab from a molecular perspective.','*8','SET','')
newCmd('.spacefill','','','*v+11.0 -- adds solvent-accessible surface "+" option;disp','Renders selected atoms as shaded spheres. A boolean value renders the spheres with the van der Waals radius. A decimal value specifies the sphere radius in Angstroms. An integer followed by "%" specifies sphere radius as a percentage of the van der Waals radius. A "+" sign followed by a number specifies to draw the surface that number of Angstroms beyond the van der Waals radius. See also {#.dots~} and {#.isosurface~isosurface sasurface}. Note that the value of the current spacefill setting can be retrieved using the .radius value for the atom. (For example, print {{atomno=3}}.radius.) [Note: The Jmol default Van der Waals radius setting is "AUTO" to allow non-PDB files and PDB files with H atoms to load with a slightly different look than PDB files with no H atoms. This brings Jmol\'s default parameter set in line with OpenBabel 2.2 and affects the default rendering also of {#.halos~} and {#.star~stars}.]','0|1','','')
newCmd('','','','','Turns spacefill on/off','*11','._on_off{"ON"}','')
newCmd('','','','*v+11.6;','Turns spacefill rendering on and all other rendering (including labels) off.','*12','ONLY','')
newCmd('','','','','The default setting, renders atom sizes for non-PDB files or PDB files with H atoms using OpenBabel 2.2 values. ','*21','AUTO','')
newCmd('','','','','Specifying a decimal number generates a sphere at a specific radius in Angstroms. A negative number also implies ONLY.','*22','[decimal]','')
newCmd('','','','','Specifying an integer percent generates a sphere at the specified percentage of the van der Waals radius. This refers to the currently set van der Waals radius set -- Jmol, Babel, Rasmol, or User.','*31','[integer]','%')
newCmd('','','','*v+11.6;vdw','Specifying an integer percent generates a sphere at the specified percentage of the van der Waals radius as defined by Jmol 10 constants (OpenBabel 1.0). See {misc/vdw_comparison.xls~vdw_comparison.xls}.','*32','[integer]','%Jmol')
newCmd('','','','*v+11.6;vdw','Specifying an integer percent generates a sphere at the specified percentage of the van der Waals radius as defined by OpenBabel 2.2. See {misc/vdw_comparison.xls~vdw_comparison.xls}','*331','[integer]','%Babel')
newCmd('','','','*v+12.0;vdw','Specifying an integer percent generates a sphere at the specified percentage of the van der Waals radius as defined by OpenBabel 2.1. See {misc/vdw_comparison.xls~vdw_comparison.xls}','*333','[integer]','%Babel21')
newCmd('','','','*v+11.6;vdw','Specifying an integer percent generates a sphere at the specified percentage of the van der Waals radius as defined by Rasmol. See {misc/vdw_comparison.xls~vdw_comparison.xls}','*34','[integer]','%Rasmol')
newCmd('','','','*v+11.6;vdw','Specifying an integer percent generates a sphere at the specified percentage of the van der Waals radius as defined by the user using the {#.data~} command.','*35','[integer]','%User')
newCmd('','','','','With an explicit plus sign, spacefill draws the surface the given number of angstroms beyond the van der Waals radius. This is the definition of a solvent-accessible surface. Note that spacefill +0 is the same as spacefill 100%. More typical would be spacefill +1.2.','*4','+(solvent probe radius)','')
newCmd('','','','','Generates a sphere for eah atom according to its crystallographic B-factor. If Uij data has been read from a CIF file, then this value is set to 100 * (U11 * U22 * U33)^0.333','*6','TEMPERATURE','')
newCmd('','','','','Generates a sphere for each atom according to an approximation of its ionic radius.','*5','IONIC','')
newCmd('','','','*v+11.6;','Generates a sphere at the radius corresponding to the minimum anisotropic displacement parameter for the selected atoms factored by the given percentage. See also {#.ellipsoid~}.','*7','ADPMIN','n%')
newCmd('','','','*v+11.6;','Generates a sphere at the radius corresponding to the maximum anisotropic displacement parameter for the selected atoms factored by the given percentage. See also {#.ellipsoid~}.','*8','ADPMAX','n%')
newCmd('.spin','','','*v+11.0 -- adds molecular frame spin/rotate and fully integrates spin and rotate as one feature;anim;rspin','Starts and stops the molecule spinning around the axis determined by {#.set_spinX~set spinX, set spinY, and set spinZ}, as though the "camera" were changing position. This command was greatly expanded in Jmol 11.0 to include all the possible functions of {#.rotate~}.','0|1','','')
newCmd('','','','*v+11.0;','','1','._on_off{"ON"}','')
newCmd('.ssbonds','','','bond','Cysteine disulfide bonds can be turned on or off, colored, and given customized widths in Angstroms.','0|1','','')
newCmd('','','','','','*1','._on_off{"ON"}','')
newCmd('','','','','','*2','._ssbond_width_angstroms','')
newCmd('','','','','','*3','._ssbond_width_rasmol','')
newCmd('.star','','','*v+11.6 -- adds new capability;','The star command places a set of crosshairs of a given size in Angstroms on the selected atoms. The default size of the star is, like spacefill, the nominal van der Waals radius for the atom. The default color for the star is that of the atom it is associated with. Use {#.color~color star [colorname]} to then color selected stars the color of your choice. For options, see {#.spacefill~}. ','1|1','','')
newCmd('.step','','','*v+11.8 -- NEW ;pause','Within the Jmol application, after a {#.pause~} command, step executes only the next command and then pauses again. To see what the next command is without executing it, enter ? as the command. ','0|1','','')
newCmd('.stereo','','','*v+11.0 -- adds customizable colors;','Jmol supports two forms of stereo rendering for molecules. In the first form, the two images are placed side by side and rotated so as to appear from slightly different perspectives, creating the illusion of 3D when a practiced user trains one eye on one image and the other eye on the other image.
','0|1','','')
newCmd('.slab','11.1.5','','*v+11.2 adds internal slabbing;slabdepth;disp;misc',' Slab and Depth together control the percentage of the molecule to be displayed based on clipping planes. All of the commands described here for {#.slab~} are also available for {#.depth~}. slab on turns slab/depth on. slab 50 shows the back 50% of the molecule. slab 25 show the back 25% of the molecule. depth does the opposite of slab, hiding atoms far from the user. The default settings to see all of the model, then, are slab 100; depth 0. Atoms appear solid; bonds appear hollow. Slabbing can also be applied "internally" -- that is, based on molecular coordinates, not screen coordinates. Internal slabbing involves defining a plane ax + by + cz + d = 0 as {{a b c d}}, using Miller indices {{h k l}}, or using standard notation such as "x=3" or "xy". CHIME NOTE: The slab/depth effect is equivalent to the RasMol command \'set slabmode solid\', however \'set slabmode [option]\' is not supported. ','0|1','','')
newCmd('','','','','Turns slab/depth on or off. Either slab ON or depth ON can be used; in either case both slab and depth are turned on.','*1','._on_off{"ON"}','')
newCmd('','','','','Sets the position of the slab/depth plane from 0 (front) to 100 (rear) of the model','*2','._percent_slab','')
newCmd('','11.1.5','','*v+11.2;','Sets the position of the slab/depth plane based on the specified Miller index plane. For slabbing, with positive hkl values, atoms far from {{0 0 0}} are removed. The value NONE removes the slab/depth plane','*32','HKL','{{h k l}} or NONE')
newCmd('','11.1.5','','*v+11.2;','Sets the position of the slab/depth plane based on the specified Miller index plane; reference point is opposite slab hkl or depth hkl.','*33','-HKL','{{h k l}}')
newCmd('','11.1.5','','*v+11.2;','Sets the slab/depth plane based on a plane using the general syntax for {#.plane expressions~}. For example: slab PLANE {{atomno=3}} {{atomno=2}} {{atomno=1}}; slab on. Note that the order of points defines the direction from the plane of atoms to be removed. If using your right hand to define the path from first point to second to third, your right thumb points to the atoms removed by slabbing. (Opposite this for depth.)','*41','PLANE','plane_expression or NONE')
newCmd('','11.1.5','','*v+11.2;','Reverses the sense of the plane defined as above.','*42','-PLANE','plane_expression')
newCmd('','11.1.5','','*v+11.2;','Resets slab and depth to the standard slab 100, depth 0, clears any internal planes, and turns slab on.','*7','RESET','')
newCmd('','11.1.5','','*v+11.2;','Sets the current slab (using slab on) to be internal, so that rotation of the model preserves the current slab from a molecular perspective.','*8','SET','')
newCmd('.spacefill','','','*v+11.0 -- adds solvent-accessible surface "+" option;disp','Renders selected atoms as shaded spheres. A boolean value renders the spheres with the van der Waals radius. A decimal value specifies the sphere radius in Angstroms. An integer followed by "%" specifies sphere radius as a percentage of the van der Waals radius. A "+" sign followed by a number specifies to draw the surface that number of Angstroms beyond the van der Waals radius. See also {#.dots~} and {#.isosurface~isosurface sasurface}. Note that the value of the current spacefill setting can be retrieved using the .radius value for the atom. (For example, print {{atomno=3}}.radius.) [Note: The Jmol default Van der Waals radius setting is "AUTO" to allow non-PDB files and PDB files with H atoms to load with a slightly different look than PDB files with no H atoms. This brings Jmol\'s default parameter set in line with OpenBabel 2.2 and affects the default rendering also of {#.halos~} and {#.star~stars}.]','0|1','','')
newCmd('','','','','Turns spacefill on/off','*11','._on_off{"ON"}','')
newCmd('','','','*v+11.6;','Turns spacefill rendering on and all other rendering (including labels) off.','*12','ONLY','')
newCmd('','','','','The default setting, renders atom sizes for non-PDB files or PDB files with H atoms using OpenBabel 2.2 values. ','*21','AUTO','')
newCmd('','','','','Specifying a decimal number generates a sphere at a specific radius in Angstroms. A negative number also implies ONLY.','*22','[decimal]','')
newCmd('','','','','Specifying an integer percent generates a sphere at the specified percentage of the van der Waals radius. This refers to the currently set van der Waals radius set -- Jmol, Babel, Rasmol, or User.','*31','[integer]','%')
newCmd('','','','*v+11.6;vdw','Specifying an integer percent generates a sphere at the specified percentage of the van der Waals radius as defined by Jmol 10 constants (OpenBabel 1.0). See {misc/vdw_comparison.xls~vdw_comparison.xls}.','*32','[integer]','%Jmol')
newCmd('','','','*v+11.6;vdw','Specifying an integer percent generates a sphere at the specified percentage of the van der Waals radius as defined by OpenBabel 2.2. See {misc/vdw_comparison.xls~vdw_comparison.xls}','*331','[integer]','%Babel')
newCmd('','','','*v+12.0;vdw','Specifying an integer percent generates a sphere at the specified percentage of the van der Waals radius as defined by OpenBabel 2.1. See {misc/vdw_comparison.xls~vdw_comparison.xls}','*333','[integer]','%Babel21')
newCmd('','','','*v+11.6;vdw','Specifying an integer percent generates a sphere at the specified percentage of the van der Waals radius as defined by Rasmol. See {misc/vdw_comparison.xls~vdw_comparison.xls}','*34','[integer]','%Rasmol')
newCmd('','','','*v+11.6;vdw','Specifying an integer percent generates a sphere at the specified percentage of the van der Waals radius as defined by the user using the {#.data~} command.','*35','[integer]','%User')
newCmd('','','','','With an explicit plus sign, spacefill draws the surface the given number of angstroms beyond the van der Waals radius. This is the definition of a solvent-accessible surface. Note that spacefill +0 is the same as spacefill 100%. More typical would be spacefill +1.2.','*4','+(solvent probe radius)','')
newCmd('','','','','Generates a sphere for eah atom according to its crystallographic B-factor. If Uij data has been read from a CIF file, then this value is set to 100 * (U11 * U22 * U33)^0.333','*6','TEMPERATURE','')
newCmd('','','','','Generates a sphere for each atom according to an approximation of its ionic radius.','*5','IONIC','')
newCmd('','','','*v+11.6;','Generates a sphere at the radius corresponding to the minimum anisotropic displacement parameter for the selected atoms factored by the given percentage. See also {#.ellipsoid~}.','*7','ADPMIN','n%')
newCmd('','','','*v+11.6;','Generates a sphere at the radius corresponding to the maximum anisotropic displacement parameter for the selected atoms factored by the given percentage. See also {#.ellipsoid~}.','*8','ADPMAX','n%')
newCmd('.spin','','','*v+11.0 -- adds molecular frame spin/rotate and fully integrates spin and rotate as one feature;anim;rspin','Starts and stops the molecule spinning around the axis determined by {#.set_spinX~set spinX, set spinY, and set spinZ}, as though the "camera" were changing position. This command was greatly expanded in Jmol 11.0 to include all the possible functions of {#.rotate~}.','0|1','','')
newCmd('','','','*v+11.0;','','1','._on_off{"ON"}','')
newCmd('.ssbonds','','','bond','Cysteine disulfide bonds can be turned on or off, colored, and given customized widths in Angstroms.','0|1','','')
newCmd('','','','','','*1','._on_off{"ON"}','')
newCmd('','','','','','*2','._ssbond_width_angstroms','')
newCmd('','','','','','*3','._ssbond_width_rasmol','')
newCmd('.star','','','*v+11.6 -- adds new capability;','The star command places a set of crosshairs of a given size in Angstroms on the selected atoms. The default size of the star is, like spacefill, the nominal van der Waals radius for the atom. The default color for the star is that of the atom it is associated with. Use {#.color~color star [colorname]} to then color selected stars the color of your choice. For options, see {#.spacefill~}. ','1|1','','')
newCmd('.step','','','*v+11.8 -- NEW ;pause','Within the Jmol application, after a {#.pause~} command, step executes only the next command and then pauses again. To see what the next command is without executing it, enter ? as the command. ','0|1','','')
newCmd('.stereo','','','*v+11.0 -- adds customizable colors;','Jmol supports two forms of stereo rendering for molecules. In the first form, the two images are placed side by side and rotated so as to appear from slightly different perspectives, creating the illusion of 3D when a practiced user trains one eye on one image and the other eye on the other image.  A second form of stereo rendering, anaglyphic rendering, nearly superimposes two identical models that are slightly rotated relative to each other. These models are each of a different color (red and one of blue, cyan, or green). The illusion of depth can then be created when the user wears an inexpensive pair of "3D glasses" that have differently colored lenses.
A second form of stereo rendering, anaglyphic rendering, nearly superimposes two identical models that are slightly rotated relative to each other. These models are each of a different color (red and one of blue, cyan, or green). The illusion of depth can then be created when the user wears an inexpensive pair of "3D glasses" that have differently colored lenses. One should experiment with different background colors when using redcyan or redblue stereo rendering. For many users background grey looks better than background white or background black.','1|2','','') newCmd('','','','','Turns side-by-side stereo viewing on. (Note that if this form of stereo viewing is desired, you will probably want to have the applet width twice the applet height.) The default rotation is -5 degrees. Sets the number of degrees of clockwise vertical-axis rotation of the RIGHT-hand image relative to the LEFT-hand image (which itself does not change rotation when stereo viewing is turned on and off). Negative values correspond to cross-eyed viewing, where the left eye is trained on the right image, and the right eye is trained on the left image. Positive values (clockwise rotation) correspond to "wall-eyed" viewing, where the right eye is trained on the right image and the left eye is trained on the left image. Note that stereo 90 may be useful, as it shows two views of a model that rotate synchronously, a "front view" on the left and a "right side view" on the right.','*1','._stereo_angle{5}','') newCmd('','','','','Turns side-by-side stereo viewing on with a previously defined rotation or, if no rotation has been defined, a rotation of 5 degrees.','*2','{"ON"}','') newCmd('','14.3.15','','*v+14.4;','Turns side-by-side DTI format stereo viewing on with a previously defined rotation or, if no rotation has been defined, a rotation of 5 degrees. The left and right eye versions are placed side by side, with the horizontal dimension halved so that both images appear in the same space of the standard display. The viewing angle is optional.','*31','DTI','._stereo_angle{5}') newCmd('','','','','Turns stereo viewing off.','*30','OFF','') newCmd('','','','','Turns red/blue anaglyphic rendering on with a specific relative rotation, if desired. The default is 3 degrees.','*4','REDBLUE','._stereo_angle{3}') newCmd('','','','','Turns red/cyan anaglyphic rendering on with a specific relative rotation, if desired. The default is 3 degrees.','*5','REDCYAN','._stereo_angle{3}') newCmd('','','','','Turns red/green anaglyphic rendering on with a specific relative rotation, if desired. The default is 3 degrees.','*6','REDGREEN','._stereo_angle{3}') newCmd('','','','*v+11.0;','Turns custom two-color anaglyphic rendering on with a specific relative rotation, if desired. The default is 3 degrees. The second color is optional. If it is left off, then the second color is the complement of the first. So, for example: stereo red is the same as stereo red cyan. Note that due to the odd way JavaScript designates green (as [x008000] rather than [x00FF00]), stereo red green is NOT the same as stereo redgreen. The equivalent of stereo redgreen is stereo red lime. When experimenting to match a given set of glasses, it is recommended that you use explicit hex codes for the colors:
stereo [xFF0000] [x00FF00]
The set of color names used in Jmol is the JavaScript set, given at {http://jmol.sourceforge.net/jscolors/#JavaScript colors~http://jmol.sourceforge.net/jscolors/#JavaScript colors}.','*7','._colorRGB','._colorRGB','._stereo_angle{3}','') newCmd('.strand','','structure','disp','Strands offer a representation the protein backbone or nucleic acid helix using lines. Curvature control points are as described for {#.ribbon~}. ','0|1','','') newCmd('','','','','','*11','._on_off{"ON"}','') newCmd('','','','*v+11.6;','Turns strand rendering on and all other rendering (including labels) off.','*12','ONLY','') newCmd('','','','','Normally, strands vary in width according to the amino acid atom positions. This command sets the radius of the set of strands to be a constant value (a decimal, in Angstroms). A negative number also implies ONLY.','*2','._strand_radius','') newCmd('.structure','','','*v+11.6 -- new;','The structure command allows customized definition of the secondary structure of a protein model as HELIX, SHEET, TURN, or NONE. ','0|1','','') newCmd('','','','','The atom expression is optional and if missing defaults to the currently selected atoms. structure HELIX {{4-10:B}}, for example, sets residues 4-10 on chain B to be represented as a helix. Note that the file must be of PDB format (from a pdb, mmcif, or suitable mol2 file) as well.','*1','HELIX|SHEET|TURN|NONE','(atomExpression)') newCmd('.struts','11.9.22','','*v+12.0 - new;bond, hbond, measure, struts','In rapid prototyping of protein models, it is sometimes necessary to add short connectors between strands and helixes to provide strength to the plastic model. Jmol includes the shape STRUT that creates these supports. These stick-like objects can be added using the {#.connect~CONNECT ... STRUTS} or {#.set picking~SET PICKING STRUTS} commands, and they can be calculated automatically using {#.calculate ~calculate STRUTS}. STRUTS are not measures and they are not covalent bonds. set strutDefaultRadius 0.3 sets the default radius for struts. (Their color by default is translucent white.)','0|1','','') newCmd('','','','','Turn struts on or off','*11','._on_off{"ON"}','') newCmd('','','','','Turns struts on and all other rendering (including labels) off.','*12','ONLY','') newCmd('','','','','Show struts with the given cylinder diameter in Angstroms','*2','._radius_angstroms','') newCmd('','','','','Show struts with the given cylinder diameter in Rasmol units (deprecated).','*3','._radius_rasmol','') newCmd('.subset','','','*v+11.0 -- new;dhide','The subset command selects a subset of the atoms that will serve as a basis for all atom expressions until either another subset command is issued or until a new file is loaded. After subset *:C, for example, select * only selects atoms of chain C, restrict none only clears chain C, and restrict *:C does nothing. By itself, the subset command is the same as subset all. Since all menu-driven commands work through the script processor, realize that any user selections will also be affected by the current subset. ({#.Hover~hover} is still active, though.) "Select All" from the menu, for example, will only select the current subset of atoms. In addition, atoms outside the current subset cannot be clicked on by the user for atom identification or for measurements. Thus, after operations requiring subset have been completed, consider issuing subset all unless you intend to shut off user clicking.','0|1','','') newCmd('.switch','','','*v+12.0 -- new;math;flow','switch and case allow a simplified syntax as an alternative to mulitple {#.if~} statements. Like JavaScript, CASE values can be string or integer constants. Unlike JavaScript, CASE values are evaluated at run time, so they can also be expressions, not just constants, as in:
x = "test"
switch("test") {{
case x::>print "OK":>break;
}}
Also, unlike JavaScript, the tests for CASE equivalence in Jmol are case insensitive, so cases "f" and "F" are indistinguishable. Expressions are evaluated from the top down; the DEFAULT keyword may appear along with any other CASE or by itself. As in JavaScript, an optional BREAK statement is necessary after a CASE to prevent continuation of the script into the next CASE. For example:
switch(prompt("What sort of rendering would you like?","Spacefill|Wireframe|Ball&Stick",true)) {{
case "spacefill"::> spacefill only:>break
case "wireframe"::> wireframe only:>break;
case "ball&stick"::>wireframe only;wireframe reset;spacefill reset:>break;
}}','0|1','','') newCmd('.sync','12.2.0','','*v+13.0 adds synchronization with JSpecView and socket-based communication (as used for the Molecular Playground);sync','The sync command allows synchronization among applets and applications. [||Applet Synchronization | For applets, an applet can be a driver, a slave, or (like many supervisors) both (a slave driver, GET IT?). Three optional settings are available. With set syncScript ON scripted commands are transmitted; with set syncMouse ON mouse movements are transmitted. These two parameters are independent. If both are off (the default behavior) and sync is ON, then the orientation of the target applet is maintained the same as the driving applet, but script commands are not automatically transmitted. Details can be seen at {examples-11/sync2.htm~}.||Inter-Application Communication|The sync command with a numerical first parameter allows Jmol to function as a server and/or client over a local internet port. In server mode (specified with a negative port number), Jmol accepts connections and then responds to JSON-encoded messages relating to script commands, mouse actions, touches, or shut-down requests. As a client, Jmol can accept such messages or also send messages to a connected server. Protocol details can be found in {misc/appsync.txt~}.||JSpecView Synchronization|Jmol incorporates {http://jspecview.sourceforge.net~JSpecView} into the application and includes special sync capabilities that allow JCAMP-DX files that contain spectra, models, and assignments for IR, NMR, MS, GC, and GC-MS data to be automatically synchronized for user interaction with the data and the model. This capability within the application extends to the applet, in which case a page would load two applets, JmolApplet.jar and JSpecViewApplet.jar. For example, see {http://chemapps.stolaf.edu/jmol/jspecview/test2.htm~http://chemapps.stolaf.edu/jmol/jspecview/test2.htm}.
The format is sync ˜ "Select: attributes" where attributes are key/value pairs in the form of HTML or XML: key="value". Attributes include file, model, atoms, select, and script. If Jmol does not find a model with ID file#model, it will load that model. atoms is a list of atom numbers separated by commas: 1,2,3, etc., that will be translated into the selection @1 or @2 or @3 within Jmol. select can be any valid Jmol selection such as THR or 1-30. Jmol automatically adds visible and to the selection created from atom or select. Requires SYNC ON. See {misc/Jmol-JSpecView-specs.pdf~Jmol-JSpecView-specs.pdf}||]','1|2','','') newCmd('','11.3.9','','*v+11.4;','Turns on synchronization for just this applet (.), all applets except this one (>), all applets (*), a specific applet on this page (appletId), or a specific applet within a specific synchronization set (appletId[syncId]). All applets selected will drive orientations of all others.','*1','&PER;|>|*|appletId|appletId[syncId]','ON') newCmd('','11.3.9','','*v+11.4;','Same as sync ON, but the selected applets will be set to receive commands only, not send them.','*2','','SLAVE') newCmd('','11.3.9','','*v+11.4;','Turns off synchronization for the designated applets.','*3','','OFF') newCmd('','11.3.9','','*v+11.4;','Sends the command (or scripted sequence of commands) to the desigated applets. Quotation marks ARE required.','*4','','"command"') newCmd('','12.2.RC8','','*v+12.2;','(application only) Indicating a negative port number initializes Jmol as a server over the specified port. Also sets sync on; sync slave.','*5','-(integer)','') newCmd('','12.2.RC8','','*v+12.2;','(application only) Acting as a server, sends a command to its own instance (primarily for testing).','*6','-(integer)','command') newCmd('','12.2.RC8','','*v+12.2;','(application only) Sends a Jmol command over the specified port, establishing the connection if it does not exist.','*7','(integer)','command') newCmd('','12.2.RC8','','*v+12.2;','(application only) Sends a JSON message to a server. (See {misc/appsync.txt~}.)','*8','(integer)','JSON message') newCmd('','14.3.11','','*v+14.4;','(application only) Sends a JSON message to a server using a Jmol variable or expression. For example:
x = {{"type":"command","command":"background green; print y", "var": "y", "data":[\'an array\',2]}}
sync 3000 x
','*9','(integer)','expression') newCmd('.throw','14.1.11','','*v+14.2;flow,pause','The throw command initiates a user-defined error condition that either stops processsing or, when within a {#.try~try/catch} syntax, is caught and handled at another point in the script. ','1|2','','') newCmd('','14.1.11','','*v+14.2;','The throw command initiates a user-defined error condition that either stops processsing or, when within a {#.try~try/catch} syntax, is caught and handled at another point in the script. For example:
:>print "testing":>throw "testing here":>print "we will never see this"
results in the following message in the Jmol console:
:>testing:>script ERROR: testing here:>----:>throw >> "testing here" <<
The message need not be just a simple string. It can be any Jmol variable or any math expression. For example:
:>x = "i=":>throw x + i
The thrown message is passed to a variable named thrown_value, which can be checked within the catch part of a try/catch syntax:
:>try{{:>>print "testing":>>throw "testing here":>>print "continuing":>}} catch(e) {{:>>print "thrown_value=" + thrown_value;:>}}:>
results in::>:>testing:>thrown_value=testing here
','*1','[math expression]','') newCmd('','14.1.11','','*v+14.2;','The throw CONTEXT form of the throw command allows an unusual form of asynchronous processing that is unlike anything in Java or JavaScript. The command replaces {#.pause~} and {#.resume~}, now deprecated. The command throws a catchable error, but in the process of doing so, creates a script context that allows RESUMING the process at the point of the throw. If within a try/catch phrase, throw CONTEXT contextName is handled by the catch, in which case the variable thrown_value is just the string contextName (and @thrown_value will be the context itself, in the form of an associative array).
More interesting, though, is when the throw is not within a try/catch phrase, in which case Jmol reports to the console:
:>to resume, enter: &contextName
This essentially provides a callback into the running (though interrupted) script. So you could, for example, put a running script "on hold" while you load a file or check or modify variables, and then continue where it left off.
Resuming the process at the point of the throw is carried using {#.resume~resume CONTEXT contextName} or just & followed by the name of the context:
:>throw context test:>...:>&test
The current context returns to the highest level, out of all {#.for~} or {#.while~} loops or {#.function~} calls. However, the variables in the context that was thrown are accessible as though the contextName variable were an associative array. So, for example, if we have
:>function f(a, b, c) {{:>>var x = 5:>>throw context testing:>>print "x=" + x:>}}
:>f(1,2,3):>print "done"
The context will be saved as the variable "testing", which takes the form of an associative array with keys that are lower-case names of all variables defined for this context (using {#.var~VAR}). We can then test all the variables in that saved context and even change them before continuing:
:>print testing["x"]
:>5
:>testing["x"]++
print testing
:>_path : [script] >> function f:>_retval : 0:>a : 2:>b : 2:>c : 3:>x : 6
If we were then to issue
:>&testing
we would get the report:
:>x=6:>done
Note that contexts can be resumed any number of times. The variables will be whatever values they were left at in the previous time through the process (not necessarily their original values.)
Note also that resuming contexts within functions that return a value, for example, in the function myfunc within
:>x = a + myfunc(a,b,c) + c
will not (at this time) return to complete the assignment. Instead, the process will continue at the next statement in the script. If this sort of functionality is desired, it is recommended that arguments be passed by reference, as arrays:
:>x = [ ]:>myfunc(a,b,c,x) // fills in x[1] rather than returning a value:>x = a + x[1] + c
It is possible that future versions of Jmol will be able to resume within an expression and complete the assignment, so throws within such functions should be avoided.
','*2','CONTEXT','contextName') newCmd('.timeout','11.9.24','','*v+12.0 - new;','Sets a script to be executed after a given time. (Execution can be checked using set debugscript. The commands as they are executed will appear in the {#.console~}.) See also {#show~show timeout}. ','2|3','','') newCmd('','','','*v+12.0;','By itself, the timeout command simply displays a list of pending timeouts.','*1','','') newCmd('','','','*v+12.0;','Turns off all pending timeouts or <no timeouts set>','*2','OFF','') newCmd('','','','*v+12.0;','Initiates a timeout. A decimal delay time (for example, 3.0) is taken to be seconds; integer time indicates milliseconds. If the delay time is negative, then the timeout will repeat until a new file is loaded or a ZAP command is given. The ID keyword is optional.','*3','ID "id"','time(s or ms)','"command"','') newCmd('','','','','Turns off the named timeout if pending. The ID keyword is optional.','*4','ID "id"','OFF') newCmd('.trace','','structure','disp','A "trace" is a smooth hermite curve through the same control points used by {#.ribbons~}.','0|1','','') newCmd('','','','','','*11','._on_off{"ON"}','') newCmd('','','','*v+11.6;','Turns trace rendering on and all other rendering (including labels) off.','*12','ONLY','') newCmd('','','','','','*2','._trace_radius','') newCmd('.translate','','','*v+11.8 -- new options;anim;camera','Moves the molecule along the specified window-based axis, X, Y, or Z. If the keyword SELECTED is added as the first parameter, then only the selected atoms are moved, and actual coordinates are changed. Otherwise, unless indicated otherwise, the TRANSLATE command just moves the viewpoint, without changing atom coordinates.','1|2','','') newCmd('','','','*v+11.8;','Moves the center of rotation to the specified value. A value of 100 will move the molecule completely out of the window. The value represents the percentage of the display window, and 0 is the window center. A value of 50 will move the center of the molecule to the edge of the window. Positive values are to the right of center for X and below center for Y. ','*1','X or Y','(decimal)') newCmd('','','','*v+11.8;','Moves the center of rotation by the specified percent of the window width (X) or height (Y). ','*2','X or Y','(decimal)','%','') newCmd('','','','*v+11.8;','Moves the center of rotation by the specified number of angstroms or nanometers at the midpoint depth of the model. . Positive values shift the model to the right for X and down for Y.','*3','X or Y','(decimal)','NM or ANGSTROMS','') newCmd('','','','*v+11.8;','Adjusts the zoom setting such that the specified percent change in field of view is effected. Positive values give the illusion of moving the model toward the user. Values that would magnify the model to the extent that less than one angstrom spans the entire window are ignored.','*4','Z','(decimal)','%','') newCmd('','','','*v+11.8;','Adjusts the zoom setting such that the specified number of angstroms or nanometers are removed from the field of view (positive value, magnification) or added to the field of view (negative value, giving the illusion of zooming out) at the midpoint depth of the model, as defined by the dimension from which zoom is referenced (based on set zoomLarge or set zoomHeight). Values that would magnify the model to the extent that less than one angstrom spans the entire window are ignored.','*5','Z','(decimal)','NM or ANGSTROMS','') newCmd('','','','','Moves the indicated atoms along the specified vector, in Angstroms, changing their actual atom coordinates. {#.fractional coordinates~Fractional coordinates} are allowed and are indicated with a "/" anywhere in any of the three coordinates. If the atom set is not included, and the SELECTED keyword is used, then the currently selected atom set is translated. ','*6','{{x y z}}','{{atomSet}}') newCmd('.translateSelected','','','*v+11.0 -- new;anim','Translates a specified subset of atoms, changing their actual coordinates. This command is equivalent to translate selected .... See {#.translate~}. ','1|2','','') newCmd('.try','','','*v+12.0 - new;flow','Surrounding a set of Jmol script with try{{...}} and following it by catch(e){{...}} allows the catching of errors within larger blocks of script. The action is the same as in JavaScript, except that the catch phrase is optional. For example, in the following, the first model is never lost, and no error message is printed in the applet window. Any variables created with the VAR keyword are local to the try or catch block.
load quartz.cif
refresh
while(true) {{ :> try {{:>> load files "caffeine.xyz" "1d.pdb":> }} catch(e) {{:>> prompt @{{"oops -- " + e}}:>> break;
}}:> prompt "File loaded successfully":> break;
}}
Note that Jmol script errors are found, not necessarily Java or JavaScript errors. For example, x =load("asdfasdf") will not throw an error, because if a Java or HTML file read error is obtained, the variable x simply is filled by that error string. For load("xxx") in particular, you can {#.throw~} your own error within the try block by inspecting its result and seeing if it is valid -- for example, using try{{ load("xxx");if(x.find("Exception")||x.find("404")) throw x;. . . }}','1|2','','') newCmd('.catch','','','*v+12.0 - new;flow','See {#.try~}.','1|2','','') newCmd('.unbind','','','*v+12.0;bind','Removes the tie between a mouse action and a Jmol action or user script. For descriptions of these, see {#.bind~}.','1|2','','') newCmd('','','','*v+12.0;','By itself, unbind returns Jmol to its default mouse action binding.','*1','','') newCmd('','','','*v+12.0;','Removes all bindings to the specified mouse action.','*2','(mouse action)','') newCmd('','','','*v+12.0;','Removes all bindings to the specified jmol action','*3','(Jmol action)','') newCmd('','','','*v+12.0;','Removes all bindings to the specified user script','*4','"script"','') newCmd('','','','*v+12.0;','Removes the specified mouse action for the specified Jmol action.','*5','(mouse action)','(Jmol action)') newCmd('','','','*v+12.0;','Removes the specified mouse action for the specified script.','*6','(mouse action)','"script"') newCmd('.undo','12.2.RC6','','*v+12.2 -- new;','(application only) same as the Undo console button -- reloads the previous state','1|2','','') newCmd('.undoMove','12.2.RC6','','*v+12.2 -- new;','same as CTRL-Z -- undoes changes to atom position when in modelKit mode.','1|2','','') newCmd('.unitcell','','','*v+11.0 -- new;axes','Turns on or off the unit cell for crystal structures, and determines its line style and line width (as a decimal number, in Angstroms).','1|2','','') newCmd('','','','*v+11.0;','Turns the unit cell on or off','*1','._on_off{"ON"}','') newCmd('','','','*v+11.0;','Sets the unit cell line diameter in Angstroms. ','*21','(decimal)','') newCmd('','14.1.16','','*v+14.2;','Sets the unit cell based on the current bound box.','*22','BOUNDBOX','') newCmd('','13.1.19','','*v+13.2;','Sets the unit cell to be centered on a given atom or coordinate. The coordinate is assumed to be in fractional coordinates already, for example {{0.5 0.5 0.5}}.','*23','CENTER','._atom_expression_or_coord') newCmd('','','','*v+11.0;','Sets the axes style to a thin dotted line.','*3','DOTTED','') newCmd('','','','*v+11.0;','Sets the origin of the unit cell to be the specified coordinate (with braces). No matter how the coorinate is written (with or without "/"), it is interpreted as a fractional coordinate. For example, unitcell {{0.5 0.5 0.5}} sets the origin of the unit cell to {{1/2, 1/2, 1/2}}. This change is for display purposes only -- the actual "{{0 0 0}}" point remains where it was initially. The keyword OFFSET, introduced in Jmol 14.2, is optional.','*4','OFFSET','{{i j k}}') newCmd('','12.2.RC6','','*v+12.2;','Creates a block of unit cells, possibly scaled, using the "555" cell naming system, where 555 is the base cell, and digits 0-9 indicate cells -5 to +4 from relative to this for each axis. A negative scale, such as -0.5, fills the specified range of unit cells with unit cells of the specified size allowing, for instance, filling of a set of supercells with unit cells based on a primitive smaller unit cell. A scale of 0 creates the unit cell but does not fill it with smaller unit cells. Starting in Jmol 14.2, more complicated supercells can be generated using the "abc;offset" method (see below). The keyword RANGE, introduced in Jmol 14.2, is optional. Jmol implements extended versions of 555 in the form of 1505050 (range 0 to 99) and 1500500500 (range 0 to 999), allowing for descriptions of a much wider range of cells. Thus, 1505152 and 1500501502 are same as 567, but 1505070, cell {{1 1 21}}, has no corresponding 555 representation.','*50','RANGE','{{nnn yyy scale}}') newCmd('','11.9.12','','*v+12.0;ticks','Sets the parameters for ticks along the a, b, and c edges of the unit cell. An optional specific axis (X, Y, or Z) can be indicated. There are three levels of ticks - major, minor, and "subminor." Only the major ticks have labels. Which of these tick levels are displayed and the distance between ticks depends upon the parameter that takes the form of a point. This point may be in fractional form, {{1/2 0 0}}. The optional keyword FORMAT allows formating of the labels for the major ticks. These are based on an array of strings given after the FORMAT keyword. If the array is shorter than the number of ticks, the formats in the array are repeated. ','*63','TICKS X|Y|Z','{{major,minor,subminor}} FORMAT ["%0.2f", ...] ') newCmd('','14.1.15','','*v+14.2;','An array of four points defines a new unit cell. Note that fractional units may be used, and if they are, then they are defined for the CURRENT unit cell. For example: unitcell [{{0 0 0}} {{1/2 0 0}}, {{0 1/1 0}} , {{0 0 1/1}}] is reversed by unitcell [{{0 0 0}} {{2/1 0 0}}, {{0 1/1 0}}, {{0 0 1/1}}]. Note that use of "/" in a ALL of these points, indicating fractional coordinates.','*640','[ {{origin}} {{a}} {{b}} {{c}} ]','') newCmd('','','','','Sets the unit cell from a string such as "a=10,b=10,c=20,alpha=90,beta=90,gamma=129".','*641','"a=...,b=...,c=...,alpha=...,beta=...,gamma=...."','') newCmd('','','','','Sets the unit cell from a 4x4 rotation-translation matrix.','*642','TRANSFORM','[4x4 matrix]') newCmd('','','','','Sets the unit cell to be its reciprocal, indicating a multiple of pi using an integer.','*52','RECIPROCAL','(integer)') newCmd('','','','','Sets the unit cell to be its reciprocal, scaling by the indicated factor.','*53','RECIPROCAL','x.x') newCmd('','14.1.16','','*v+14.2;','Sets the unit cell to be the parent unit cell (MCIF reader only)','*65','"parent"','') newCmd('','14.1.16','','*v+14.2;','Sets the unit cell to be the stanadard unit cell (MCIF reader only)','*66','"standard"','') newCmd('','14.1.16','','*v+14.2;','Sets the unit cell to be the conventional (file-based) unit cell. If the unit cell from the file is primitive, you can use UNITCELL @{{unitcell("conventional")}} to generate a conventional cell from it.','*674','"conventional"','') newCmd('','14.4.4','','*v+14.6;','Sets the unit cell to be a primitive unit cell','*678','"primitive"','') newCmd('','14.1.16','','*v+14.2;','Sets the unit cell to a new setting relative to its current setting. abc is an expression such as a/2,2b,c that relates the new setting to the current setting. offset is an optional fractional offset such as 0,1/2,0 relative to the current unit cell. Adding "!" at the beginning of the description reverses the notation. Thus, unitcell "!a,2b,c;0,1/2,0" reverses the effect of unitcell "a,2b,c;0,1/2,0". This command can be used to create completely different unit cells from that given in a file and allowing commands such as select cell=555 or draw unitcell or print {{Fe1}}.fxyz to operate on this new unit cell.','*68','"abc;offset"','') newCmd('','14.1.16','','*v+14.2;','Returns the unit cell to its original setting and resets its line width to a thin line. (Note that {#.restore~restore unitcell} accomplishes the same without resetting the line width.)','*51','RESET','') newCmd('.unset','12.1.32','','*v+12.2;rreset','Same as {#.reset~}; just looks better for variables. Good to use if just a small part of a large array or hashtable has been assigned to another variable and the rest can be discarded.','1|2','','') newCmd('.var','','','flow','The var keyword is used with variable definitions to localize them to a specific script file or {#.functions~function}. Note that var variables within {#.for~}, {#.while~}, {#.try~}, and {#.try~catch} blocks or any code surrounded by {{ }} except {#.if~} and {#.switch~} blocks. Starting with Jmol 14.2, as in JavaScript, var can be used with multiple variable names: var x,y,z. (Jmol does not support defining variables when listed this way as is possible in JavaScript.)','1|2','','') newCmd('.vector','','','disp','Draws vectors arising from vibrational mode data. Operates on whichever atoms have been selected. See also {#.color (atom object)~}. For msCIF or JANA2006 incommensurately modulated structures , turning "vectors on" displays an arrow from the unmodulated position to the modulated position when {#.modulation~} is off, and the other direction when modulation is on. ','1|2','','') newCmd('','','','','Turns vibration vector display on and off','*1','._on_off{"ON"}','') newCmd('','','','','Sets the diameter of the vector','*2','._diameter_pixels','') newCmd('','','','','Sets the diameter of the vector','*3','._radius_angstroms','') newCmd('','','','','Adjusts the scale of the vector independently of the vibration motion.','*4','SCALE','._vector_scale') newCmd('.vibration','','','*v+11.0 adds vibration period command;vib','Provided information is present in the file (xyz format with columns 6-8 indicating dx, dy, and dz, or Gaussian harmonic frequency output), turns on and off display of vibration animation and allows setting of the time period for the vibration (in seconds) and the scale of the motion relative to the default (1). For msCIF or JANA2006 incommensurately modulated structures , turning "vibration on" actuates a visualization of the modulations as totally nonrealistic (but still valuable) phased vibrations.','1|2','','') newCmd('','','','','Turns vibration on with a 1-second time period or turns vibration off.','*1','._on_off{"ON"}','') newCmd('','','','','Sets the time period for one full vibration in seconds and turns vibration on. ','*20','._time_period','') newCmd('','','','*v+14.4;','Rescales all vibration vectors to have a maximum value specified.','*21','MAX','(decimal)') newCmd('','','','*v+11.0;','Sets the time period for one full vibration in seconds, but does not turn vibration on','*3','PERIOD','._time_period') newCmd('','','','','Sets the scale of the vibration motion independently of the vector length.','*4','SCALE','._vibration_scale') newCmd('.while','','','*v+11.4 -- new flow control options;math;flow','while, like {#.if~} evaluates a logical expression, looping until the expression is no longer TRUE:
while (x > 0 && x < 10) {{:>print x:>x = x - 2
}}
The looping can be discontinued using either {#.break~} or {#.break~continue}. Any variables created with the VAR keyword are local to the while block.','1|2','','') newCmd('.wireframe','','','disp;bond','Wireframe refers to the bonds drawn between the atoms. A boolean value of ON draws the selected bonds as lines. Alternatively, a numeric value may be used to specify the radius of the bonds. A decimal value such as 0.2 or 0.4 specifies Angstroms. The wireframe command operates on bonds either BETWEEN ANY TWO atoms in a previously selected atom set (having previously {#.setbondstyles~set bondmode AND}) or TO ANY atoms in a previously selected atom set (having previously {#.setbondstyles~set bondmode OR}). Note that the selected atoms must already be connected (based on information in the file, Jmol\'s autobonding algorithm, or from use of the {#.connect~} or {#.bondorder~} command) in order to show any effect. CHIME NOTE: Rasmol and Chime will take wireframe 0 as a one-pixel-width bond (equal to wireframe and hence to wireframe on), while Jmol will interpret it as wireframe off.','0|1','','') newCmd('','','','','Turn wireframe on or off','*11','._on_off{"ON"}','') newCmd('','','','*v+11.6;','Turns wireframe rendering on and all other rendering (including labels) off.','*12','ONLY','') newCmd('','','','','Show wireframe with the given cylinder diameter in Angstroms. A negative number also implies ONLY.','*2','._radius_angstroms','') newCmd('','','','','Show wireframe with the given cylinder diameter in Rasmol units (deprecated).','*3','._radius_rasmol','') newCmd('.write','','','*v+13.0 adds fully encapsulated PNG files ("PNGJ" format);clip','(application or HTML5 or signed Java applet only). Writes information to a file or to the system clipboard (or POSTing to a server using a URL "http://..." for the file name. Options include: an image of the application screen, the script history, the current model state in the form of a script, the current molecular orbital in the form of a JVXL file, or the current isosurface in the form of a JVXL file. If the filename is simple (no spaces) and the type is clearly in the extension as, for example, write test.spt, then Jmol will deduce the type from the filename and create the proper file. For the clipboard, simply specify CLIPBOARD instead of a file name. For files created with zip formats (PNGJ, JMOL, ZIP), the command show FILE "<filename>" will list the files contained in the ZIP files. The write() function can be used to place the output of the write command into a variable or to the console. For example: x = write("coord", "MOL") or x = write("PNGJ").','0|1','','') newCmd('.write','','','*v+12.0 adds LOCALPATH and REMOTEPATH options for write STATE;*v-13.0;clip','(application or signed applet only). Writes information to a file or to the system clipboard (or POSTing to a server using a URL "http://..." for the file name. Options include: an image of the application screen, the script history, the current model state in the form of a script, the current molecular orbital in the form of a JVXL file, or the current isosurface in the form of a JVXL file. If the filename is simple (no spaces) and the type is clearly in the extension as, for example, write test.spt, then Jmol will deduce the type from the filename and create the proper file. For the clipboard, simply specify CLIPBOARD instead of a file name. For files created with zip formats (PNGJ, JMOL, ZIP), the command show FILE "<filename>" will list the files contained in the ZIP files.','0|1','','') newCmd('','14.1.9','','*v+14.2;','Starting with Jmol 14.2, you can also specify the type after the filename and the AS keyword: write test.png as PNGJ. The AS keyword only allows this simple format -- just a filename and a type. If any more parameters are required, the AS keyword cannot be used. The type may be any of the file types described below.','*1','"fileName"','AS','type','') newCmd('.write (export)','','','*v+11.8 adds IDTF, VRML, and X3D options;','Jmol models can be exported to several formats that can be read by external rendering programs. ','0|1','','') newCmd('','11.8.RC5','','*v+11.8;','Exports the current scene as an {http://u3d.svn.sourceforge.net/viewvc/u3d/trunk/Docs/IntermediateFormat/IDTF%20Format%20Description.pdf~Intermediate File} that can be easily converted to a {http://en.wikipedia.org/wiki/Universal_3D~Universal 3D} file for incorporation into 3D-PDF documents. The conversion to U3D is done external to Jmol using the (Windows-only) program {misc/idtf.zip~}. (This is a copy from the Bin/Win32/Release directory found in the zip file downloadable from {http://sourceforge.net/projects/u3d/~}; source code for that C++ program is available at this site as well.) Once Jmol has created the IDTF file using, for example, write IDTF "myfile.idtf", simply execute from a Windows command window IDTFConverter.exe -input myfile.idtf -output myfile.u3d. The U3D file created will be highly compressed and FAR smaller than that created using other means (such as first creating a VRML file and then converting that to the U3D format). ','*1','IDTF','"fileName"') newCmd('','','','*v+11.4;','Exports the current scene as a Maya file. (Basic elements such as atoms and bonds only.)','*2','MAYA','"fileName"') newCmd('','','','*v+12.2;','Exports the current scene as a WaveFront OBJ file. The file "fileName" is written along with the additional material file, "fileName.mtl".','*34','OBJ','"fileName"') newCmd('','','','*v+11.8;','Exports the current scene as POVRAY. The file "fileName" is written along with the additional file, "fileName.ini". (Typically "fileName" would include the ".pov" extension, so these would be of the form xxx.pov and xxx.pov.ini.) Note that the .pov file will embed the Jmol script, so the model can be loaded back into Jmol using {#.script~script "xxx.pov"}.','*35','POVRAY','"fileName"') newCmd('','12.1.26','','*v+12.2;','Creates a {http://www.symyx.com/downloads/public/ctfile/ctfile.pdf~V3000} file.','*41','V3000','"fileName"') newCmd('','','','*v+11.4;','Exports the current scene as VRML.','*42','VRML','"fileName"') newCmd('','11.8.RC4','','*v+11.8;','Exports the current scene as X3D.','*5','X3D','"fileName"') newCmd('','14.8','','*v+11.8;','Exports the current scene as WRL for 3D printers. See the {#.struts~} command for adding additional printed supports.','*45','WRL','"fileName"') newCmd('','14.8','','*v+11.8;','Exports the current scene as STL for 3D printers. STL is primarily a binary format. The ASCII format will be produced when debugging is turned on using set debug TRUE. See the {#.struts~} command for adding additional printed supports.','*37','STL','"fileName"') newCmd('.write (images and frames)','','','*v+11.0;image','Jmol can write a 2D image of the model and, for JPG and PNG format, can append its state so that a single file can be read either as an image or as a Jmol state. In addition, You can write sequences of frames as a set of JPG files that can then be incorporated by other programs into movie files. Image quality is determined by {#.set_antialiasImages~set antialiasImages}, which is distinct from {#.set_antialiasDisplay~set antialiasDisplay}. ','0|1','','') newCmd('','11.7.1','writeframes','*v+11.8;','Allows making of Jmol movies, creating a set of JPG files filename0001.jpg, filename0002.jpg, etc. for all frames in the atom expression. These files can then be combined using an external program of user\'s choice to create a movie from a sequence of JPEG images. Width and height are optional, as is the atom expression.','*111','FRAMES','.atom_expression','width','height "fileName.jpg"') newCmd('','12.3.28','writeframes','*v+12.4;','Creates a sequence of JPG file frames that can be combined with movie-making software into a movie. The number of vibration cycles, n, determines the number of frames (individual JPG files) that will be created (20 * n).','*112','VIBRATION','n','"fileName"','') newCmd('','','','*v+11.0;','Creates a "snapshot" image of the current display and saves it to disk as a GIF image. width and height are optional and default to the current frame size. Note that when many colors are present, Jmol may not produce satisfactory GIF images.','*121','IMAGE width height','GIF','"fileName"','') newCmd('','14.3.8','','*v+14.4;','Creates a "snapshot" image of the current display and saves it to disk as a transparent-background GIF image. width and height are optional and default to the current frame size. It is recommended that the background color [x040404] be used for window, labels, echo, and scale for best transparency.','*122','IMAGE width height','GIFT','"fileName"','') newCmd('','','','*v+11.0;','Creates a "snapshot" image of the current display and saves it to disk as a JPG image. width and height are optional and default to the current frame size. The optional integer n is a quality from 1 to 100 (default 100; or 75 prior to Jmol 13.1.15). If set imageState is TRUE (the default setting), then Jmol inserts into the JPG file its state, allowing a single image file to be used both for viewing as a 2D image and reading as a 3D Jmol state using the {#.script~} command or by dragging and dropping in the Jmol application.','*123','IMAGE width height','JPG','n','"fileName"') newCmd('','','','*v+11.0;','Creates a "snapshot" image of the current display and saves it to disk as a JPG or base-64-encoded JPG image. width and height are optional and default to the current frame size. The optional integer n is a quality from 1 to 100 (default 75).','*14','IMAGE width height','JPG64','n','"fileName"') newCmd('','13.3.9','','*v+11.0;','Creates a "snapshot" image of the current display embedded in a PDF file and saves it to disk. width and height are optional and default to the current frame size. The optional parameter n specifies portrait (1, default) or landscape (2) mode. ','*151','IMAGE width height','PDF','n','"fileName"') newCmd('','','','*v+11.0;','Creates a "snapshot" image of the current display in PNG format and saves it to disk. width and height are optional and default to the current frame size. The amount of compression, n, is a number between 0 and 10 (default 2). If set imageState is TRUE (the default setting), then Jmol appends to the PNG file its state, allowing a single image file to be used both for viewing as a 2D image and reading as a 3D Jmol state using the {#.script~} command or by dragging and dropping in the Jmol application. For this drag/drop operation to be successful, any local files necessary (model, or isosurface, for example) must be in the directory containing the PNG file, and any remote files must still be accessible via a web connection. See also the PNGJ format, below.','*152','IMAGE width height','PNG','n','"fileName"') newCmd('','12.3.7','','*v+13.0;','Creates a "snapshot" image of the current display in PNG format, appending to that file the full state and all necessary files (models, isosurfaces, etc.) required to reproduce it in the form of JMOL zip data format. width and height are optional and default to the current frame size. Note that the zip-data appendix in a PNGJ file can be inspected using x = load("test.png") (keys only) or x = load("test.png", true) (key/value pairs) and written out as a ZIP file using x = load("test.png",true); write var x "test.zip".','*16','IMAGE width height','PNGJ','n','"fileName"') newCmd('','12.2.RC6','','*v+12.2;','Creates a "snapshot" image of the current display in PNG format, setting the background to be transparent. It is recommended using an unusual background color, such as [x101010], to ensure only the background is made transparent. Also, set antialiasImages FALSE is recommended to avoid ragged edges around the image. width and height are optional and default to the current frame size. The amount of compression, n is a number between 0 and 10 (default 2). ','*17','IMAGE width height','PNGT','n','"fileName"') newCmd('','','','*v+11.0;','Creates a "snapshot" image of the current display in {http://netpbm.sourceforge.net/doc/ppm.html~PPM} format and saves it to disk. width and height are optional and default to the current frame size. ','*18','IMAGE width height','PPM','"fileName"','') newCmd('.write (info)','','','*v+11.0;','The Jmol history, menu, currently defined functions, and variables may all be written to files. In addition, specific properities, quaternion analysis, and ramachandran information can also be saved in PDB format.','0|1','','') newCmd('','11.3.23','','*v+11.4;','Writes all user-defined functions to a file. ','*12','FUNCTIONS','"fileName"') newCmd('','','','*v+11.0;','Saves the script command {#.history~} to a file.','*20','HISTORY','"fileName"') newCmd('','11.9.4','','*v+12.0;','Same as ZIPALL (see below). See also write PNGJ, above.','*3','JMOL','"fileName"') newCmd('','','','*v+11.8;','Saves the current Jmol popup menu as a file. See {#.load MENU~}.','*4','MENU','"fileName"') newCmd('','','','*v+12.0;','Writes a file that is of PDB format and contains the quaternion representation of the rotational frame of individual amino acid residues in a protein or base pairs in a nucleic acid. See the {#.plot~plot QUATERNION} command for parameter options.','*5','QUATERNION','. . . . ','"fileName"','') newCmd('','','','*v+12.0;','Writes a file that is of PDB format and contains the specified parameters in the X, Y, and Z fields, possibly scaled, with the scaling indicated in a REMARK record. See the {#.plot~plot PROPERTIES} command for parameter options.','*71','PROPERTIES','. . . . ','"fileName"','') newCmd('','','','*v+14.4;','Writes a file with the format you specify. For example: write properties x y z format "%s %i %f %f %f" "t.dat". The fields %s and %i refer to (optional) atom name and index, respectively.','*73','PROPERTIES (list) FORMAT "%s %i . . ."','"fileName"') newCmd('','','','*v+12.0;','Creates a file in PDB format for the alpha carbons of a peptide, where the x, y, and z axis values are actually the PHI, PSI, and OMEGA values for each amino acid. An additional optional keyword DRAW writes a script file that would draw PHI and PSI angles on the structure. See the {#.plot~plot RAMACHANDRAN} command for parameter options.','*8','RAMACHANDRAN','. . . . ','"fileName"','') newCmd('','13.1.2','','*v+13.2;','Writes the specified data directly to a file.','*21','INLINE','"data"','"fileName"','') newCmd('','11.1.14','','*v+11.2;','Saves the value of a variable to a file. If x is a binary associative array containing the key "_IMAGE_", then a PNGJ file is created. If x is a binary associative array NOT containing this key, then a ZIP file is created. Note that the zip-data appendix in a PNGJ file can be written out as a ZIP file using the following command: x = load("test.png",true); write var x "test.zip".','*9','VAR','[variable name]','"fileName"','') newCmd('.write (model)','','','*v+11.2;','There are several options for writing model data to a file or saving the state of Jmol so that a model can be reloaded. ','0|1','','') newCmd('','11.1.13','','*v+11.2;','Writes molecular data to a file either determined by keyword or file extenion. The type keyword may be omitted if the filename has an letter extension matching one of these types. Only the currently selected atoms are saved.MOL67 creates MOL files that utilizes the "aromatic single/double" bond types 6 and 7. XYZRN creates files that include van der Waals radius and name, for input to the {http://www.scripps.edu/˜sanner/html/msms_home.html~MSMS} program. XYZVIB creates a multi-model XYZ file with vibrational vectors added for all atoms having defined vibration vectors. The SDF option creates a multi-model {http://www.symyx.com/downloads/public/ctfile/ctfile.pdf~SDfile}. Jmol automatically adds a ><JMOL_ATOM_NAMES> section. You can add to user data by assigning key/value pairs in the model\'s molData property. For example,
:>molData = {{"atom_values":"chirality"}}:>model properties "molData" molData:>write test.sdf
Note that if the model already has a molData property, then use the following to append to it:
:>molData = (_M.molData ? _M.molData : {{}}):>molData.atom_values = "chirality":>model properties "molData" molData
','*1','CIF|CML|CFI|MOL|PDB|PQR|SDF|CD (ChemDoodle)|JSON|QCJSON|V2000|V3000|XYZ|XYZRN|XYZVIB','"fileName"') newCmd('','11.1.13','','*v+11.2;','Writes a script file explicitly containing the coordinates of the previously selected atoms, whether or not they have been altered using {#.invertSelected~}, {#.rotateSelected~}, or {#.translateSelected~}. The SPT keyword is optional if the file ends with the extension .spt.','*22','COORDS','SPT','"fileName"','') newCmd('','','','','Writes a file including only the previously selected atoms, modifying the coordinates to the current view if the file type is one of: CD, JSON, MOL, SDF, V2000, or V3000. Starting with Jmol 14.2, if the filename is omitted then the coordinates in XYZ format are displayed in the console.','*21','COORDS','"fileName"') newCmd('','11.3.39','','*v+11.4;','Writes the most recent file loaded to the indicated filename. If the model was loaded using a manifest from a ZIP file, then the entire ZIP file is written.','*3','FILE','"fileName"') newCmd('','11.9.4','','*v+12.0;','Same as ZIPALL (see below). See also write PNGJ, above. The JMOL keyword is optional if the file ends with the extension .jmol.','*4','JMOL','"fileName"') newCmd('','','','*v+11.0;','Saves the current state to a script file. The STATE keyword is optional if the file ends with the extension .spt. When a state is loaded, the stateVersion variable is set. This variable can be checked to see what version of Jmol was used to create the state:
write "t3.spt";script "t3.spt";print stateVersion
1403015 # indicating Jmol 14.3.15
','*5','STATE','"fileName"') newCmd('','11.9.2','','*v+12.0;','Saves the current state to a script file, stripping all local file references to the indicated relative path. ','*6','STATE','LOCALPATH','"path"','"fileName"') newCmd('','11.9.2','','*v+12.0;','Saves the current state to a script file, stripping all remote file references to the indicated relative path. ','*7','STATE','REMOTEPATH','"path"','"fileName"') newCmd('','11.9.4','','*v+12.0;','Saves the current state and all necessary LOCAL files to a ZIP file with the given name. The ZIP keyword is optional if the file ends with the extension .zip.','*8','ZIP','"fileName"') newCmd('','11.9.4','','*v+12.0;','Saves the current state and all necessary files -- local or remote -- to a ZIP file with the given name. The ZIPALL keyword is optional if the file ends with the extension .jmol.','*9','ZIPALL','"fileName"') newCmd('.write (object)','','','*v+11.2;','Several Jmol objects can be saved independently.','0|1','','') newCmd('','','','*v+11.2;','Saves the current isosurface in the form of a JVXL file. Specifying the file extension as .pmesh or .pmb creates an ASCII (.pmesh) or binary (.pmb) file in a Jmol-specific file format. This can speed up loading of meshes and contours, but in general the default .jvxl format is recommended. ','*1','ISOSURFACE','"fileName"') newCmd('','11.9.23','','*v+12.0;','Generates a relatively compact JVXL XML "vertex-only" mesh surface file from an isosurface. A typical command, after creation of an isosurface, would be write MESH t.jvxl. Note that standard JVXL files are considerably smaller, however.','*2','MESH','"fileName"') newCmd('','','','*v+11.2;','Saves the current molecular orbital in the form of a JVXL file.','*3','MO','"fileName"') newCmd('','','','*v+11.6;pg','Writes a tabular summary of the point group for a symmetrical or nearly symmetrical molecule. ','*4','POINTGROUP','"fileName"') newCmd('','','','*v+11.6;pg','Creates a script file containing DRAW commands that will draw the planes and axes of symmetry for a molecule. ','*5','POINTGROUP','DRAW','"fileName"','') newCmd('.zap','','','*v+11.6 -- selective model removal;;load;rreset','Clears the currently loaded molecule. After zap or before any model is loaded, all of the following commands are active and may be used to create nonmolecular visualizations: {#.axes~}, {#.background~}, {#.boundbox~}, {#.center~}*, {#.centerAt~}, {#.dipole~}*, {#.draw~}*, {#.echo~}, {#.font~}, {#.frank~}, {#.isosurface~}*, {#.move~}, {#.moveto~}, {#.pmesh~}, {#.restore~}, {#.rotate~}*, {#.save~}, {#.slab~}, {#.spin~}*, {#.stereo~}, {#.translate~}, and related {#.set~} commands.
* indicates only aspects not requiring selection of atoms for these commands. The ZAP command can be used to delete selected models.','1','','') newCmd('','','','*v+11.6;','Clears all loaded models.','*1','','') newCmd('','','','*v+11.6;','Clears all models that include any of the selected atoms. Examples include: zap file=1, zap model=1.1, zap atomno=1, zap not visible','*2','(atom expression)','') newCmd('.zoom','','','*v+11.0 -- new capabilities;anim',' Allows enlarging or shrinking of the displayed model, as though the "camera" were moving toward or away from the model. A percentage value specifies the zoom relative to 100, the default value, which in Jmol is calculated so that all atoms are completely visible on the screen (when {#.set_zoomlarge~set zoomLarge} is set FALSE), or visible in the largest dimension (when {#.set_zoomlarge~set zoomLarge} is set TRUE, its default setting), through all rotations using the default vanderWaals rendering percentage. The command "zoom OFF" disables mouse-based zooming and zooms to 100. The command "zoom ON" re-enables zooming at the current zoom setting. If the zoom has been turned off, setting the zoom using, for example, "zoom 50," though it sets the "current zoom setting," has no effect until the next "zoom ON" command is given. If an atom expression or isosurface ID is given, then zoom also sets the center of that object as the rotation center, and if windowCentered is true (the default), that point is moved the center of the screen. ','*1','._on_off{"ON"}','') newCmd('','','','*v+12.2;','Zooms to the setting that fills the screen with the currently displayed atoms.','*21','0','') newCmd('','','','*v+12.0;','Zooms IN by a factor of 2 over the course of 1 second.','*15','IN','') newCmd('','','','*v+12.0;','Zooms OUT by a factor of 2 over the course of 1 second.','*16','OUT','') newCmd('','','','','Zooms to the specified percent zoom','*20','._percent_zoom','') newCmd('','','','*v+11.0;','Sets a new zoom setting and optionally also designates the specified reference atom expression or coordinate or isosurface as the rotation center. If a reference atom set or isosurface is given, the value is relative to that group of atoms or isosurface.','*3','optional {{atom expression}} or {{x y z}} or $xxx','percent zoom or 0') newCmd('','','','*v+11.0;','Adds or subtracts an absolute amount from the current zoom setting and optionally sets the rotation center.','*4','optional {{atom expression}} or {{x y z}} or $xxx',' + or - delta') newCmd('','','','*v+11.0;','Multiplies or divides the current zoom setting by the indicated factor and optionally sets the rotation center.','*5','optional {{atom expression}} or {{x y z}} or $xxx',' * or / factor') newCmd('.zoomto','','','*v+11.0 -- new;anim;nav','Carries out a smooth transition to the specified zoom setting. Indicating a new rotation center is optional. All capabilities of {#.zoom~} are supported. Jmol includes centering on isosurfaces using $xxx.','1|0','','') newCmd('','','','*v+11.0;','By itself, zoomTo smoothly zooms IN by a factor of 2 over the course of 1 second.','*1','','') newCmd('','','','*v+12.0;','Smoothly zooms IN by a factor of 2 over the course of 1 second.','*21','IN','') newCmd('','','','*v+11.0;','Smoothly zooms OUT by a factor of 2 over the course of 1 second.','*22','OUT','') newCmd('','','','*v+11.0;','Smoothly moves the specified atom or coordinate to the center of the window if windowCentered or designates it as the center of rotation if not windowCentered. xTrans and yTrans parameters specify percent translation within the window along X and Y, respectively . All parameters are options. The default time is 1 second; indicating no center position results in simple, smooth zooming.','*3','._floattime','{{atom expression}} or {{x y z}} or $xxx') newCmd('','','','*v+11.0;','Smoothly transitions to the indicated zoom setting over the course of the specified number of seconds. Optional xTrans and yTrans parameters specify percent translation within the window along X and Y, respectively ','*4','._floattime','{{atom expression}} or {{x y z}} or $xxx','._percent_zoom','xTrans yTrans') newCmd('','','','*v+11.0;','Adds or subtracts an absolute amount from the current zoom setting over the course of the specified number of seconds. Optional xTrans and yTrans parameters specify percent translation within the window along X and Y, respectively ','*5','._floattime','{{atom expression}} or {{x y z}} or $xxx',' + or - delta','xTrans yTrans') newCmd('','','','*v+11.0;','Multiplies or divides the current zoom setting by the indicated factor over the course of the specified number of seconds. Optional xTrans and yTrans parameters specify percent translation within the window along X and Y, respectively ','*6','._floattime','{{atom expression}} or {{x y z}} or $xxx',' * or / factor','xTrans yTrans') newCmd('','11.1.36','','*v+11.2;','Zooms to the setting that fills the screen with the designated atoms. (Requires set perspectiveModel 11 for proper operation.) Can include modifiers +n, -n, *n, /n after the 0. Optional xTrans and yTrans parameters specify percent translation within the window along X and Y, respectively ','*7','._floattime','{{atom expression}} or {{x y z}} or $xxx','0','xTrans yTrans')