Click on a link below to see what it does. You can also type a command in the box below the model to see its effect. The script run in this case was load caffeine.mol.
This page lists all new features in Jmol 12.3 (which, when released, will be 12.4). Features new to Jmol 12.2 (with links there to previous versions) are described in new2.htm Documentation for all commands can be found in the Interactive Documentation for Jmol 12.4. For the complete change log development summary, see Jmol.properties. Comments? Suggestions? Bob Hanson
Be sure you have the Latest version. Very latest may appear as Jmol-12.zip prior to release at SourceForge.
Jmol 12.3.26 adds fully validated MMFF94 molecular mechanics minimization. If no atoms are selected, the minimization is carried out on the current model; otherwise only specific atoms are minimized. (This is unchanged; see the minimization command for full details.) Any time the MMFF94 atom types cannot be found, for example if the model does not have multiple bonding indicated, the minimization reverts to UFF (Universal Force Field). The default maximum minimization steps is 100, but the minimization can be continued by using MINIMIZE again.
Jmol 12.3.26 adds the capability to run a script with parameters. One simply adds a set of variables in parentheses after the script file name and then within the script those values will be found in the _arguments array. On this site, the file testsf.spt simply reads prompt _arguments. the command name SCRIPT is optional if the file name is in quotations or ends with ".spt".
Jmol 12.3.26 adds an ID for models. This allows going to a specific model by name and is used as part of the syncronization between the Jmol and JSpecView applets. IDs are not case-sensitive.
31. measures "2:%VALUE %UNITS//xx and set defaultDistanceLabel "%UNITS//xx"
Jmol 12.3.26 adds the capability to specify units for a given distance measurement, a set of selected distance measurements, or all future distance measurements to a specific unit such as angstroms or nanometers
Jmol 12.3.26 allows setting the minimization force field to either MMFF94 or UFF. The default is MMFF, but Jmol automatically switches to UFF if atom types cannot be set. Any minimizationCallback and the variable _minimizationForceField both report the actual force field used.
Jmol 12.3.24 allows posting information to a server using the write command. The information is sent as application/octet-stream and can be an image or JMOL zip file or a Jmol variable. The return from the server can be used as an internal Jmol AJAX communication (but better!).
Jmol 12.3.24 adds easy PubChem searching using compound names, CAS numbers, PubChem CID numbers, or smiles strings. This capability requires the signed applet. click here to load the signed applet. Please be patient for this server to respond.
Jmol 12.3.22 adds a completely new API that allows pages showing Jmol applets to show either images or ChemDoodle canvases instead, depending upon the capabilities of the device showing the page. Demonstration pages include simple2.htm. The new API also provides considerably more flexibility for the Jmol applet itself, irrespective of ChemDoodle. The JavaScript is object-based and deprecates Jmol.js.
Jmol 12.3.22 adds the capability to write a model to ChemDoodle JSON format. For example: java -jar JmolData.jar -iRJ "load $tylenol;print write('cd')" generates: {"mol":{"a":[{"x":0.20549999,"y":0.8303,"z":0.3823},{"x":0.6906,"y":-1.4656999,"z":-0.14220001},...]}}
Jmol 12.3.21 fully implements headless Java operation specifically designed for server-side operation. The -R (restricted) flag allows the server to restrict operation to no reading or writing of local (server-side) files (other than a final image file using the -g and -w flags). The -T (timeout) flag sets the number of seconds to allow before the operation is canceled. The best file to use on the server is JmolData.jar, which has no graphics, and the preferred call looks something like this: java -Djava.awt.headless=true -Xmx512m -jar "JmolData.jar" -RJ "load $tylenol;" -g1000x1000 -wJPG:t.jpg. Headless operation disables command threading and the DELAY, TIMEOUT, PAUSE, LOOP, GOTO, SPIN [rate], and ANIMATION ON commands, and turns SPIN [rate] [degrees] into just ROTATE [degrees].
Jmol 12.3.19 allows setting of a model by an atom set. The first atom in the set determines which model is displayed. For example, model {within(smarts,"[r3]")} would bring into frame the first model with a 3-membered ring.
Jmol 12.3.16 marks a new collaboration with Robert Lancashire and the JSpecView project. with the incorporation of JSpecView into the Jmol application. This is most evident in that there is a new menu item Tools...JSpecView, which opens a new JSpecView frame that includes the current model. Work is in progress to bring this interface to fruition, involving the design and implementation of a new JCAMP-DX format that incorporates models and assignments into JDX files using custom ##$MODELS and ##$PEAKS records. Progress can be monitored here. the Jmol application command SYNC ON; sync * "JSpecView:..." sends commands to JSpecView.
Jmol 12.3.15 adds an additional method of interaction between applets. The sync command option Select: was specifically designed for interaction with the JSpecView applet, and has attributes much like an XML tag including file, model, atoms, select, and script. If Jmol does not find a model with ID file#model, it will load that model. atoms is a list of atom numbers separated by commas: 1,2,3 that will be translated into the selection @1 or @2 or @3. select can be any valid Jmol selection such as THR or 1-30. Jmol automatically adds visible and to the selection created from atom or select. Requires SYNC ON.
Jmol 12.3.11 allows loading of models in reverse order. This is particularly useful for developing animations of IRC calculation where the second part of the pathway may need to be reversed when combined with the first.
Jmol 12.3.10 adds a powerful new frame alignment option that is particularly useful for multimodel files where you want to align several models with one reference model. This is useful for IRC (intrinsic reaction coordinate) calculations and particularly helpful in patching together reaction mechanism sequences from different files. The default syntax is simply COMPARE {from} {to} FRAMES where {from} is a set of frames and {to} is the reference frame. All of the options of the COMPARE command, such as SUBSET, ATOMS, ROTATE, and TRANSLATE are still available. (ROTATE and TRANSLATE are implied by the FRAME keyword if neither is given.) If {to} is a subset of {from} -- for example, COMPARE {*} {1.1}, then the FRAMES keyword is unnecessary. See the mp directory for more examples.
Jmol 12.3.10 adds a way of finding out (approximately) how long it will take for an animation to run once through. This allows a script to wait for an animation to run a specific number of times and then continue. It was needed for the St. Olaf installation of its Molecular Playground.
Jmol 12.3.9 adds a way of pausing a running animation at any specific frame. The delay applies to all models specified in the most recent FRAME command.
Jmol 12.3.8 adds the capability to write a single file that can be displayed (and placed into Google documents, for example) as a PNG file but also contains all the files necessary to load the state back into Jmol. Use write PNGJ "filename" to create these files. Go ahead and copy the image on the right to your hard drive and then open it with Jmol.
The Z-Matrix format allows for molecules to be described by internal coordinates (distances, angles, and dihedrals) rather than in terms of actual Cartesian coordinates. Jmol 12.3.8 can read several forms of Z-matrix data, including Gaussan/MOPAC format as well as a format designed for Jmol that allows also the simple introduction of bond order. Details are given in the Z-Matrix reader code itself. Examples can be found in the data directory under "zmat".
Jmol 12.3.3 adds the capability to display a predicted 1H NMR spectrum. This uses the service at http://www.nmrdb.org/predictor and requires the signed applet. click here to load the signed applet.
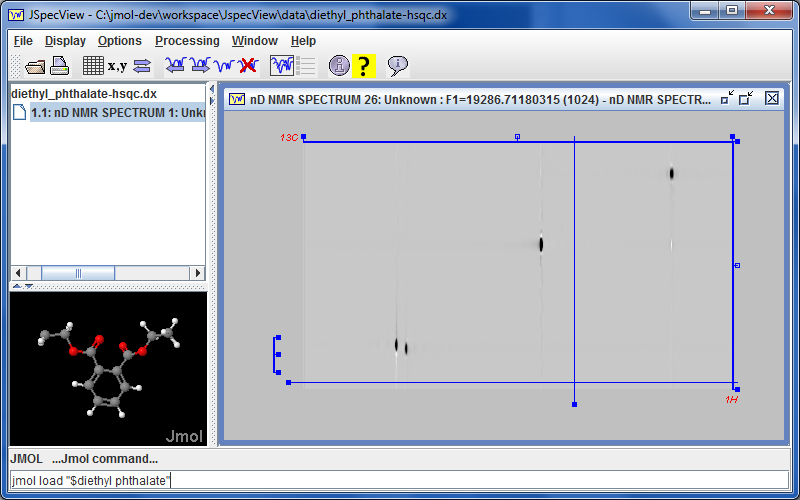 marks a new collaboration with Robert Lancashire and the
marks a new collaboration with Robert Lancashire and the  The Jmol 12.3.10 application animation play forward/reverse buttons now allow continuous animation by holding the button down.
The Jmol 12.3.10 application animation play forward/reverse buttons now allow continuous animation by holding the button down.
